Chemical databases come in many different types and flavours, and given that we now have UniChem up and running, and it is being actively used by at least some of you, our minds have turned a little to describing these ‘flavours’ and ‘resolutions’. One of the key things a user is interested in is how easy is it to get hold of a compound, since this is usually a key filter applied to actually doing anything with the results of a database search. The cost implications of needing to commission synthesis, or potentially try and develop new synthetic methodology to a compound are substantial, and there is a substantial literature on the computational assessment of synthetic accessibility (q.v.).
So, here is a simple five state classification that reflects the typical availability of a compounds in a chemical collection.
A compound has been previously been synthesized and is readily available from chemical vendors.
A compound has been previously synthesized but would require resynthesis.
A compound has not been previously synthesized, but close analogues have and the compound is likely to be readily synthesizable. This class of molecule is often associated with the phrase ‘virtual library’.
A compound has not been previously synthesized, and effort would be required to think about synthetic access to the compound.
A compound is theoretically possible with respect to valence rules, but is so unstable that it is unlikely that it ever can be isolated in pure form and then experiments in a biofluid carried out.
Of course, all these classifications are interesting, but you can do a lot more, a lot quicker and cheaper if a compound is in set 1.
As an estimate of the likely difference in cost between these different classes, I personally, would rate the cost differences, relative to set 1, as twenty fold for set 2, forty fold for set 3, and two hundred fold for set 4 - but these are just my estimates, and there will be a big variance in these costs dependent of the exact compound, its class, etc. Others will have better or different estimates of the average cost differences between the sets (comments welcome!).
Because of the way that people have assembled chemical databases, entire primary databases tend to cluster in a similar way - for example ChEMBL is mostly 2), DrugBank is mostly 1) and GDB-17 is mostly 4). Directly from the above definition, every compound with a known bioactivity has to have been synthesized, and so ChEMBL will always be a 2) in this classification. Of course, some compounds in ChEMBL are readily available, but it is a clear minority.
When people build federated chemical databases (those with little unique primary content, but primarily add value by bringing lots of feeder databases together; for example PubChem and ChemSpider) the picture gets a little more complicated at a database level, since they are often blends of synthesized and ‘virtual’ compound sets. But the same need to indicate the availability/provenance of a structure is useful, and federated databases need to store the original primary database (which may or may not itself be available outside of the federated database).
So, a couple of thoughts:
Is this classification useful to apply to the contents of UniChem?
Is the following classification of the UniChem component databases useful and valid?
One of our bff's are the guys and gals at the SGC in Oxford, they have recently got a grant to establish a University-wide Chemical Biology Platform, and are now looking for a Project Manager for this effort.
The diXa project is looking to recruit a someone to provide part-time maternity cover for the Scientific Training &
Dissemination Officer in the Industry Support team which is based at the
European Bioinformatics Institute (EMBL-EBI) located in Hinxton, near Cambridge in the UK.
The diXa project is dedicated to developing and implementing
a robust and sustainable service infrastructure for data from EU-funded
research into non-animal tests for predicting chemical safety
(http://www.diXa-fp7.eu/). EMBL-EBI is a partner in the diXa project with a
number of responsibilities.
The overall objectives for the position will be to deliver a
core set of project-related training and dissemination services to the
Toxicogenomics Research Community and to help foster effective use of resources
provided through the project working with colleagues at EMBL-EBI and the other
partner sites. The successful candidate will also liaise with other groups
involved in training and dissemination both at EMBL-EBI and at other training
partner sites. Training activities will have a strong industry focus but will
also include other stakeholders.
The key responsibilities for the position and the desired
qualifications and experience of the applicant can be reviewed on the diXa
project website.
On July 12th 2013 the FDA approved GilotrifTM (USAN afatinib) for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) carrying EGFR exon 19 deletions or exon 21 (L858R) mutation. It is a covalent, irreversible inhibitor of EGFR, ERBB2 and ERBB3.
Non small cell lung cancer (NCLS) (CRUK NCLS; PDQ NCLS) accounts for 78% of lung cancer incidences in the UK and is typically resistant to chemotherapy. In clinical studies, afatinib increased the mean Progression Free Survival to 11.1 months from 6.9 months (compared to standard of care Pemetrexed/Cisplatin). Furthermore, response to treatment was observed in tumors harboring several EGFR mutant species although the duration of response varied from 6.9 months to 16.5 months depending on the mutation .
Afatinib covalently binds to the kinase domains of EGFR (ErbB1, Uniprot: P00533; canSAR: EGFR”), HER2 (ErbB2, Uniprot:P04626; canSAR: ERBB2), and HER4 (ErbB4, Uniprot:Q15303; canSAR: ERBB4)) and irreversibly inhibits tyrosine kinase autophosphorylation, resulting in downregulation of ErbB signaling. The structure of afatinib bound to EGFR is shown (PDB=4G5J).
GilotrifTM tablets contain the dimaleate salt of afatinib. Afatinib (research code: BIBW-2992; ChEMBL id: CHEMBL1173655; SMILES:CN(C)C\C=C\C(=O)Nc1cc2c(Nc3ccc(F)c(Cl)c3)ncnc2cc1O[C@H]4CCOC4;) has a molecular weight of 485.9 and an AlogP of 3.92. The elimination half-life of afatinib is 37 hours after repeat dosing.
Registration is now open for an SMR meeting that I'll be presenting at this October. The meeting is at the National Heart and Lung Institute (NHLI) in Kensington, London. I'll be giving a talk on the work we've done on curating kinase activity data, some analysis of clinical attrition, and then mapping this to opportunities in neglected disease target space.
This was so popular last time, that I thought I would do it again. Here are some recently disclosed kinase inhibitors, which have just got assigned INN names. The competition is to assign a research code to each of these.
Answers in the comment section, please!
Update - Check out the comments for the likely research codes and sources for these - well done Jeremy!
One of the advantages (erm?) of my long distance daily five hour odd commute, is the constant company of all the imaginary friends I have on twitter (and of course Roman ;) ). Yesterday, a link to www.quantconnect.com came up in the stream, randomly; so I checked checked for a 3G signal, and browsed to the site. It is a really interesting take on a SAAS/cloud/big data business model. They've built an API, backed it with data and simulation approaches, as a way to develop and select novel trading strategies. There is also the availability of capital to execute these strategies, and for everyone to then make a profit. Smart future 'quants' in their parents spare room, with a laptop and a bunch of ideas and time can become rich, maybe. Traders at the investment banks could make some money on the side, maybe.
Anyway, have a look, it looks really nice, and got me thinking that it would be a great model for a virtual screening/drug repositioning informatics biotech, where the platform 'brokers' a tournament of approaches coded against data held on a data/compute cloud (bioactivity, patents, protein structures, models, etc). Depending on the validation results - customers (large pharma) could then apply these virtual screening strategies against their compound files, and everyone - the coder, the pharma, the patient and the platform developer are happy, maybe.


 The diXa project is looking to recruit a someone to provide part-time maternity cover for the Scientific Training & Dissemination Officer in the Industry Support team which is based at the European Bioinformatics Institute (EMBL-EBI) located in Hinxton, near Cambridge in the UK.The diXa project is dedicated to developing and implementing a robust and sustainable service infrastructure for data from EU-funded research into non-animal tests for predicting chemical safety (http://www.diXa-fp7.eu/). EMBL-EBI is a partner in the diXa project with a number of responsibilities.The overall objectives for the position will be to deliver a core set of project-related training and dissemination services to the Toxicogenomics Research Community and to help foster effective use of resources provided through the project working with colleagues at EMBL-EBI and the other partner sites. The successful candidate will also liaise with other groups involved in training and dissemination both at EMBL-EBI and at other training partner sites. Training activities will have a strong industry focus but will also include other stakeholders.The key responsibilities for the position and the desired qualifications and experience of the applicant can be reviewed on the diXa project website.Please apply online through www.embl.org/jobsThe application deadline is 29th July 2013.
The diXa project is looking to recruit a someone to provide part-time maternity cover for the Scientific Training & Dissemination Officer in the Industry Support team which is based at the European Bioinformatics Institute (EMBL-EBI) located in Hinxton, near Cambridge in the UK.The diXa project is dedicated to developing and implementing a robust and sustainable service infrastructure for data from EU-funded research into non-animal tests for predicting chemical safety (http://www.diXa-fp7.eu/). EMBL-EBI is a partner in the diXa project with a number of responsibilities.The overall objectives for the position will be to deliver a core set of project-related training and dissemination services to the Toxicogenomics Research Community and to help foster effective use of resources provided through the project working with colleagues at EMBL-EBI and the other partner sites. The successful candidate will also liaise with other groups involved in training and dissemination both at EMBL-EBI and at other training partner sites. Training activities will have a strong industry focus but will also include other stakeholders.The key responsibilities for the position and the desired qualifications and experience of the applicant can be reviewed on the diXa project website.Please apply online through www.embl.org/jobsThe application deadline is 29th July 2013.
 Afatinib covalently binds to the kinase domains of EGFR (ErbB1, Uniprot: P00533; canSAR: EGFR”), HER2 (ErbB2, Uniprot:P04626; canSAR: ERBB2), and HER4 (ErbB4, Uniprot:Q15303; canSAR: ERBB4)) and irreversibly inhibits tyrosine kinase autophosphorylation, resulting in downregulation of ErbB signaling. The structure of afatinib bound to EGFR is shown (PDB=4G5J).
Afatinib covalently binds to the kinase domains of EGFR (ErbB1, Uniprot: P00533; canSAR: EGFR”), HER2 (ErbB2, Uniprot:P04626; canSAR: ERBB2), and HER4 (ErbB4, Uniprot:Q15303; canSAR: ERBB4)) and irreversibly inhibits tyrosine kinase autophosphorylation, resulting in downregulation of ErbB signaling. The structure of afatinib bound to EGFR is shown (PDB=4G5J).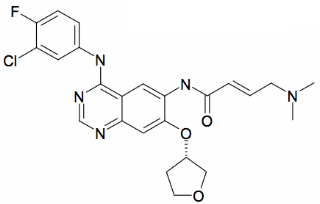

 This was so popular last time, that I thought I would do it again. Here are some recently disclosed kinase inhibitors, which have just got assigned INN names. The competition is to assign a research code to each of these.Answers in the comment section, please!Update - Check out the comments for the likely research codes and sources for these - well done Jeremy!PanulisibRiviciclibVoruciblib
This was so popular last time, that I thought I would do it again. Here are some recently disclosed kinase inhibitors, which have just got assigned INN names. The competition is to assign a research code to each of these.Answers in the comment section, please!Update - Check out the comments for the likely research codes and sources for these - well done Jeremy!PanulisibRiviciclibVoruciblib








