-
Finding Redox Pairs of Compounds

A quick question for our readers. Do you know of any online resources that maintain explicit links between redox pairs of compounds?
What I'm interested in doing is establishing links between molecules that can exist in different redox states (and can be interconverted in vivo, but are distinct in vitro) and then being able to link bioactivity data at these two redox levels. The sort of thing that's found for molecule such as NADPH/NADP+, coenzyme Q10 (ubiquinone, ubiquinol), paracetamol/NAPQI and also involved in the mechanism of action of some bioactives such as paraquat - which is an example of a viologen (which is a cool word!).
These redox pairs, of course, are not tautomers, and so won't be equivalenced by standard InChIs, but they are an important class of "equivalent" molecules, and capturing this higher level of equivalency in a comprehensive way could have big paybacks in terms of metabolomic and toxicology research.
So, if anyone knows of a list of known redox pairs they could point to or donate that would be great, and also if there are any general chemoinformatics methods to infer/enumerate alternate reasonable redox forms I'd be most interested in any pointers.
jpo -
New kinase inhibition full matrix dataset
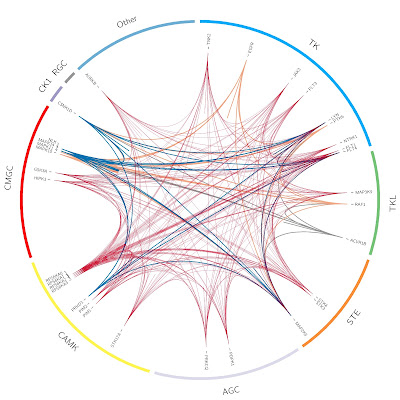 We've just processed and integrated a new full matrix kinase inhibition dataset, kindly provided by EMD Millipore. As described in this recent paper, it covers 234 human wildtype kinases and kinase complexes, as well as 158 'standard' kinase inhibitors tested at two concentrations, leading to more than 73,000 data points. Analysis of the data could provide new insights to kinase selectivity/promiscuity/polypharmacology (depends on how you look at it) and their relationship with chemical structure. The dataset will be available with ChEMBL_16 next month.PS: The image above shows the activity profile of four kinase inhibitors across the human kinome families from the Millipore data. It was produced with circos by Rita.George
We've just processed and integrated a new full matrix kinase inhibition dataset, kindly provided by EMD Millipore. As described in this recent paper, it covers 234 human wildtype kinases and kinase complexes, as well as 158 'standard' kinase inhibitors tested at two concentrations, leading to more than 73,000 data points. Analysis of the data could provide new insights to kinase selectivity/promiscuity/polypharmacology (depends on how you look at it) and their relationship with chemical structure. The dataset will be available with ChEMBL_16 next month.PS: The image above shows the activity profile of four kinase inhibitors across the human kinome families from the Millipore data. It was produced with circos by Rita.George -
Meeting up at the ACS in New Orleans

A couple of us are out in New Orleans for the ACS in a few weeks - if any ChEMBL users would like to meet up to get some training, ask questions, suggest improvements to what we do, or just grab a beer or coffee, we'd be very happy to do so. We arrive Sunday and leave Tuesday night.
The picture is rather amusing, and looks a little like Mark and me, sort of. Credit for image is detailed in the image itself.....
jpo and mark -
"I am not a number, I am a free man" - but I do have an ORCID ID!

For those of you of a certain age and interests the "I am not a number, I am a free man" phrase will mean a lot, to others nothing. Of course, on the internet you are never more than someone else's UID, but sometimes it's important to publicly make yourself unique.
I have a "rare" name, relatively - but even so some confusion is possible - there's the John Overington who is a politician in the States, and even in the scientific literature some confusion is possible with a new researcher Jeff Overington. e.g.
Overington, Jeff, et al. "Cysteine Proteases in Supernatants From Colonic Biopsies Obtained From Diarrhea-Predominant IBS Patients Evoke Sustained Hyperexcitability of Colonic Dorsal Root Ganglia Neurons." Gastroenterology140.5 (2011): S-523.
If you have a common name, like Li, for example - it's easy to get lost in the crowd.
To potentially solve this problem the ORCID ID is an attempt to give researchers a unique and persistent identifier for publications and other related stuff (like grants, patents, awards, and so on) that will survive change of name due to marriage, divorce, just 'cos you fancy a new name, or maybe even witness protection programs. However, the orcid website sums up the aims a lot more coherently....
ORCID provides a persistent digital identifier that distinguishes you from every other researcher and, through integration in key research workflows such as manuscript and grant submission, supports automated linkages between you and your professional activities ensuring that your work is recognized.
This page lists the ORCIDs of the members of the ChEMBL Team and Overington Research Group - not everyone has one at the moment, but those that do are listed here - we'll try and remember to keep this page up to date.
0000-0002-5859-1064 is in 'real' life John P. Overington.
0000-0003-0717-1817 is in 'real' life Gerard Van Westen.
0000-0002-8314-7061 is in 'real' life George Papadatos
0000-0002-8011-0300 is in 'real' life Mark Davies
0000-0002-6054-5622 is in 'real' life Felix Kruger
0000-0002-4196-475X is in 'real' life Nathan Dedman
0000-0002-5431-4756 is in 'real' life Rita Santos
jpo -
Paper: Brain: Biomedical Knowledge Manipulation

There's a paper just out from Samuel, one of the PhD students in the group - the link to the Open Access paper is here. Brain is a Java software library facilitating the manipulation and creation of ontologies and knowledge bases represented with the Web Ontology Language (OWL).
%A S. Croset %A J.P. Overington %A D. Rebholz-Schuhman %D 2013 %T Brain: Biomedical Knowledge Manipulation %J Bioinformatics %V 29 %O DOI:10.1093/bioinformatics/btt109 %O PMID:23505292
-
New Drug Approvals 2013 - Pt. IV - Ospemifene (OSPHENA®)

ATC Code: Not AssignedWikipedia: Ospemifene
On February 26, FDA approved Ospemifene (Trade Name: OSPHENA, PubChem: CID 3036505, ChEMBL: CHEMBL2105395, ChemSpider: 2300501) for the treatment of moderate to severe Dyspareunia - symptom of vulvular and vaginal atrophy due to menopause.Dyspareunia, is pain during or after sexual intercourse. It can affect men, but is significantly more common in women, affecting up to one-fifth of women at some point in their lives. Women with dyspareunia may have pain in the vagina, clitoris or labia. This may be due to medical or psychological causes. There are numerous medical causes of Dyspareunia, like : Vaginismus, Pelvic Inflammatory Disease, Genital or Pelvic Tumors, Urethritis, Urinary Tract Infection, Vaginal Atrophy, Vaginal Dryness, Vulvar Cancer, Childbirth Trauma (postpartum), Skin Conditions (Lichen Sclerosus, Lichen Planus, Eczema, Psoriasis), Female Genital Mutilation, Endometriosis - many of which can be treatable.
 Ospemifene is a novel selective estrogen receptor modulator (SERM) - class of compounds that acts on Estrogen Receptors (ER's). SERM's has a distinguishing characteristic that makes them different from pure receptor agonists and antagonists, which is - that their mode of action is different in various tissues, thereby granting the possibility to selectively inhibit or stimulate estrogen-like action in various tissues (Pub-Med). Ospemifene is an Estrogen agonist/antagonist with tissue selective effects. Its biological actions are mediated through binding to Estrogen Receptors (Short Name: ER, ESR; UniProt: Q92731 and P03372; ChEMBL: CHEMBL2093866). This binding results in activation of estrogenic pathways in some tissues (agonism) and blockade of estrogenic pathways in others (antagonism).
Ospemifene is a novel selective estrogen receptor modulator (SERM) - class of compounds that acts on Estrogen Receptors (ER's). SERM's has a distinguishing characteristic that makes them different from pure receptor agonists and antagonists, which is - that their mode of action is different in various tissues, thereby granting the possibility to selectively inhibit or stimulate estrogen-like action in various tissues (Pub-Med). Ospemifene is an Estrogen agonist/antagonist with tissue selective effects. Its biological actions are mediated through binding to Estrogen Receptors (Short Name: ER, ESR; UniProt: Q92731 and P03372; ChEMBL: CHEMBL2093866). This binding results in activation of estrogenic pathways in some tissues (agonism) and blockade of estrogenic pathways in others (antagonism).
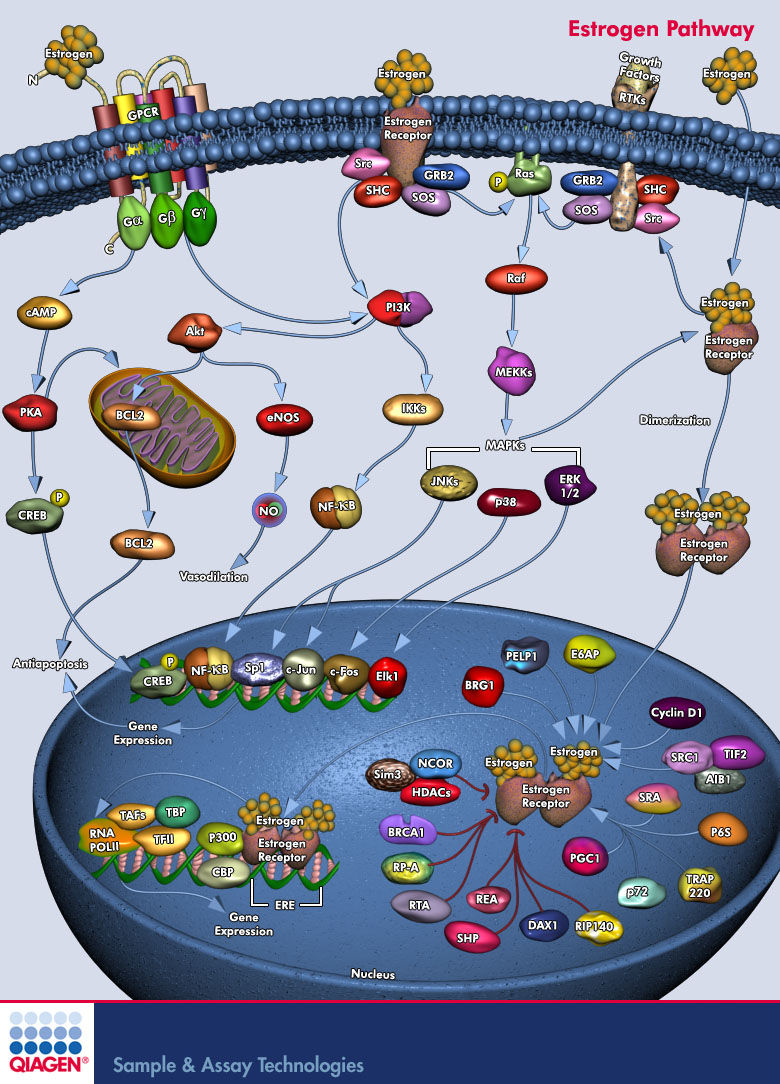
Mechanism of action of SERM's is of mixed agonism/antagonism which may differ depending on the chemical structure, but, for at least for some SERM's, it appears to be related to -1. The ratio of co-activator to co-repressor proteins in different cell types.2. The conformation of the estrogen receptor induced by drug binding, which in turn determines how strongly the drug/receptor complex recruits co-activators (resulting in an agonist response) relative to co-repressors (resulting in antagonism).
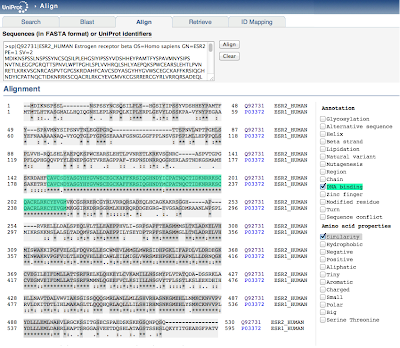
The protein sequences of human ER-alpha (ESR1) and ER-beta (ESR2) can be downloaded in fasta format from the link here. (courtesy UniProt)
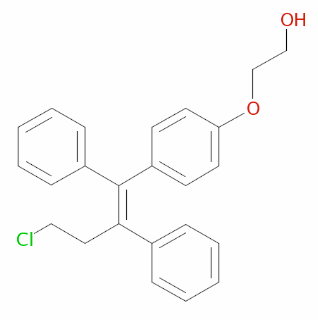 Compound Name : Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanolCanonical SMILES : OCCOc1ccc(cc1)\C(=C(\CCCl)/c2ccccc2)\c3ccccc3Standard InChI : InChI=1S/C24H23ClO2/c25-16-15-23(19-7-3-1-4-8-19)24(20-9-5-2-6-10-20)21-11-13-22(14-12-21)27-18-17-26/h1-14,26H,15-18H2/b24-23-Standard InChI Key : LUMKNAVTFCDUIE-VHXPQNKSSA-NOspemifene, an ER agonist/antagonist, has a molecular weight of 378.9. The recommended dosage is 60 mg, available in the form of table for oral administration. After single dosage of Ospemifene under fasted conditions, mean Cmax and AUC (0 to infinity) were 533 ng/mL and 4165 ng.hr/mL, respectively. With a high fat/high diet, mean Cmax and AUC (0 to infinity) were 1198 ng/mL and 7521 ng.hr/mL, respectively. It is highly bound to serum proteins ( >99%) with apparent volume of distribution of 448 L. Ospemifene primarily undergoes metabolism via CYP3A4, CYP2C9 and CYP2C19 and the major metabolite was 4-hydroxyospemifene. Clearance of ospemifene was 9.16 L/hr and terminal half-life was 26 hrs. Following an oral administration of ospemifene, approximately 75% and 7% of the dose was excreted in feces and urine, respectively.Osphena comes with a boxed warning in the form of Endometrial Cancer and Cardiovascular Disorders. Since Ospemifene is an ER agonist/antagonist with tissue selective effects. In endometrium, due to Ospemifene agonistic effects, there is an increased risk of endometrial cancer in a woman with a uterus who uses unopposed estrogens. Adding a progestin to estrogen therapy reduces the risk of endometrial hyperplasia, which may be a precursor to endometrial cancer. There is a reported increased risk of stroke and deep vein thrombosis in postmenopausal women who received daily oral conjugated estrogens alone therapy over 7.1 years. So, Ospemifene should be prescribed for the shortest duration consistent with treatment goals.Full prescribing information can be found here.The license holder is Shionogi Inc., and the product website is www.osphena.com.
Compound Name : Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanolCanonical SMILES : OCCOc1ccc(cc1)\C(=C(\CCCl)/c2ccccc2)\c3ccccc3Standard InChI : InChI=1S/C24H23ClO2/c25-16-15-23(19-7-3-1-4-8-19)24(20-9-5-2-6-10-20)21-11-13-22(14-12-21)27-18-17-26/h1-14,26H,15-18H2/b24-23-Standard InChI Key : LUMKNAVTFCDUIE-VHXPQNKSSA-NOspemifene, an ER agonist/antagonist, has a molecular weight of 378.9. The recommended dosage is 60 mg, available in the form of table for oral administration. After single dosage of Ospemifene under fasted conditions, mean Cmax and AUC (0 to infinity) were 533 ng/mL and 4165 ng.hr/mL, respectively. With a high fat/high diet, mean Cmax and AUC (0 to infinity) were 1198 ng/mL and 7521 ng.hr/mL, respectively. It is highly bound to serum proteins ( >99%) with apparent volume of distribution of 448 L. Ospemifene primarily undergoes metabolism via CYP3A4, CYP2C9 and CYP2C19 and the major metabolite was 4-hydroxyospemifene. Clearance of ospemifene was 9.16 L/hr and terminal half-life was 26 hrs. Following an oral administration of ospemifene, approximately 75% and 7% of the dose was excreted in feces and urine, respectively.Osphena comes with a boxed warning in the form of Endometrial Cancer and Cardiovascular Disorders. Since Ospemifene is an ER agonist/antagonist with tissue selective effects. In endometrium, due to Ospemifene agonistic effects, there is an increased risk of endometrial cancer in a woman with a uterus who uses unopposed estrogens. Adding a progestin to estrogen therapy reduces the risk of endometrial hyperplasia, which may be a precursor to endometrial cancer. There is a reported increased risk of stroke and deep vein thrombosis in postmenopausal women who received daily oral conjugated estrogens alone therapy over 7.1 years. So, Ospemifene should be prescribed for the shortest duration consistent with treatment goals.Full prescribing information can be found here.The license holder is Shionogi Inc., and the product website is www.osphena.com. -
How do you find ChEMBL-like papers in PubMed?

If only....
Here at ChEMBL Towers, we appreciate the delight that comes with computationally solving problems, in particular those that would otherwise be a tedious manual task - the world would be perfect if we could somehow automate and expand the search for med chem SAR literature to be included in ChEMBL by some sort of classification mechanism that distinguishes between 'ChEMBL-like papers' and 'other literature' ...
As it turns out, this type of classification problem is already addressed in other fields and is generally known as text mining. It should come as no surprise that text mining was the internal trend of the month - last month. Text mining has a lot of potential for us in ChEMBL. Possible application areas include: patent text-mining, drug repurposing, and even assay auto extraction and curation. Text mining techniques have previously been applied for in many fields, including sentiment analysis, social media mining etc.
Why not?...However, in order to tune the text mining song to our needs we decided to input some tricks from cheminformatics. The initial goal of our exercise was to create a reliable classification measure to separate 'interesting' ChEMBL papers from the rest. To this end we started with the full set of paper abstracts that are included in ChEMBL_15 (about 45,000 papers), this was our set of actives (chembl-like). In order to discriminate against another type of papers we also compiled a random subset (again about 45,000) retrieved from MedLine, this was our set of inactives (background). To capture this information we applied the Bag-of-Words (BoW) method which was constructed from both abstracts and titles.1. From the abstracts and titles punctuation and non-alphanumeric characters were removed2. The abstracts and titles were split on spaces and all words were placed in an array3. From this array uni-grams, bi-grams and tri-grams were created (eg. 'SAR', 'structure-activity relationship' or 'series was synthesized' ).4. From the n-grams stopwords were removed if they formed the majority of the n-gram.Now the sets looked like this graphically if we create a nice word cloud (ChEMBL on the left and medline on the right, previously posted by JPO - since he steals all our good stuff, and pretends it is his).
As this method quickly leads to a very large collection of possibly not so relevant words we decided to perform Bayesian feature selection. Hence the n-grams were placed in a large array and a Bayesian classifier was used to select 128 relevant n-grams (...yes we also tried 64, 256, 768, 999 or all...) that were relevant to model the classes. Keeping to chemoinformatics traditions the selected n-grams were converted into a counts-string which was used as a descriptor for each paper (...yes, we also tried bits... ).
Subsequently, the collections of bitstrings were fed into a random forest (RF) classifier (shown in our hands to outperform a Bayesian method) that was used to train a classifier. We used both out-of-bag validation and external validation (50 % training - 50 % validation). Now the nice thing about doing this with RFs is that you can see what features (in this case words) are important to distinguish ('W__' and '__' can be ignored, you'll note that we need to do some stemming too!). It seems 'compounds' is essential...

As it turns out that this type of classifier performs rather well on the very homogenous set that makes up ChEMBL. Our models consistently achieved a sensitivity of 0.91 an specificity of about 0.94 with MCC about 0.85.

So let's make it more interesting...So now that we have this method we can try some interesting exercises. We decided to train a model on ChEMBL-10 and use that to try and predict if the papers included exclusively in release 15 would be correctly classified, and in fact it did, but with a little lower accuracy (Sensitivity 0.81, specificity 0.94 and MCC 0.76). So this would mean that, while the method works, these models are not perfect.
However, we have previously decided to abstract from a relatively small set of SAR data rich journals, and have maintained this selection over time, so this result is no big deal. The real proof of concept however is a validation on completely different papers. Those that our not part of the normal ChEMBL body but might still contain interesting information to be abstracted. Here we used a set of papers (556) that have been included in BindingDB but are not in ChEMBL. The goal is to see what fraction of papers is actually retrieved by our text mining model. In total 513 of the 550 are actually flagged as ChEMBL-like (corresponding to a sensitivity of about 0.92).
However, the results should still be taken with caution, as the majority of the ChEMBL papers are from J. Med. Chem. (A) and Bioorg. Med. Chem. Lett. (B). These display a different profile than for instance the papers from J. Nat. Prod. (C, see below for the 10 top words for each of these). Still the term 'compounds' appears in all (and was also essential in our model further up). But this also points to opportunities to develop similar pipelines, sitting off PubMed updates looking for Natural Product bioactivity containing literature.In conclusion...In summary, we are very happy with this approach to document classification as it significantly increases our productivity. Currently this a approach is applied in our in house curation interface (more on this in the future, but it is cool), an ongoing project of Rita Santos (more on this later, but it is cool), and the identification of protein-family-like (e.g. GPCR-related) publications.
If you think this is interesting work please get in touch! We would be more than happy to share the PP protocols / KNIME workflows developed in house and more of our experiences doing this exercise.
Gerard & George -
The results are in - inorganics are out!

A few weeks ago we ran a small poll on how we should deal with inorganic molecules - not just simple sodium salts, but things like organoplatinums, and other compounds with dative bonds, unusual electronic states, etc. The results from you were clear, there was little interest in having a lot of our curation time spent on these. We will continue to collect structures from the source journals, and they will be in the full database, but we won't try and curate the structures, or display them in the interface. They will be appropriately flagged, and nothing will get lost. So there it is, democracy in action.
So for ChEMBL 16 expect fewer issues when you try and load our structures in your own pipelines and systems.
Thanks for all your input.
jpo












