-
Self-assembled inhibitors as an approach to PPI modulators - When Two Become One

It's a new year, so lots of planning to do, internship projects, PhD projects, etc. - so here's an idea, and you, dearest readers, can say if it's been done, or just plain stupid.
It's clear that in order to tackle new protein classes, especially for a more general approach towards protein-protein interactions (PPIs), that getting inhibitors with larger molecular weight into the body, and stable once there, is a big challenge. There's a lot of recent chatter on Teh Interwebs about the staped peptide approach, which ay well nt be as promising as once hoped. We know that there are some approximate limits to molecular weight, but to be able to get reliably up to a 800-900 MW range would and maintain good drug-like properties be a great thing. So what about achieving this with two co-dosed 400 MW inhibitors that "self assemble" in the target site to the fully effective ligand - just like in vivo fragment soaking (and just like the title of the Spice Girl's song above). The simplest way to think of achieving this would be through a targeted interaction between the two inhibitor halves, either via a direct interaction (but this will be of probable very low ligand efficiency and specificity) or maybe more interestingly - something like a specifically coordinated metal ion - like Zinc for example - there are many known biological system zinc coordinating ligands - but the simplest and most widely used is an imidazole.
%A B.A. Katz %A J.M. Clark %A J.S. Finer-Moore %A T.E. Jenkins %A C.R. Johnson %A M.J. Ross %A C. Luong %A W.R. Moore %A R.M. Stroud %T Design of potent selective zinc-mediated serine protease inhibitors %J Nature %V 391 %P 608-612 %O http://dx.doi.org/10.1038/35422

In the mid 90's there was a lot of excitement in Pharma about zinc-mediated interactions with proteinases - involving a histidine in the target protein - I remember clearly the buzz around Arris' "Delta Technology". There was a Nature publication on this (reference above), and although it was clearly a real and interesting effect, it seemed to die as a drug discovery approach - my guess is because of the very controlled and very low "free" zinc ion concentration in vivo. So this is an example of a self assembled inhibitor - anyone aware of any other examples, theoretical approaches, etc. ?
Of course, there is the issue of safety and developability for two different agents, and of any DDI's between them, and of the temporal matching of the PK profiles, but as a strategy, is it sound, and are there some nice systems to try it on? -
Paper: ChEMBLspace – a graphical explorer of the chemogenomic space covered by the ChEMBL database
 In the likely case one of your New Year resolutions was to use ChEMBL data more, here's a new paper introducing ChEMBLspace by some of our Eli Lilly collaborators. ChEMBLspace is a standalone Java application that enables users to visualise and explore an extensive network of human proteins. Two such proteins are connected if they have at least one ligand in common, as reported in the ChEMBL database (version 14). A user may select proteins and list their ligands interactively and then design an activity profile iteratively by adding more targets or anti-targets to the selection and adjusting protein-specific activity thresholds. The compounds that meet the desired profile are displayed within the application (with links to ChEMBL) and the full collection can be saved as an SD file.You may even modify the code provided and so link the application to your own in-house private data source.
In the likely case one of your New Year resolutions was to use ChEMBL data more, here's a new paper introducing ChEMBLspace by some of our Eli Lilly collaborators. ChEMBLspace is a standalone Java application that enables users to visualise and explore an extensive network of human proteins. Two such proteins are connected if they have at least one ligand in common, as reported in the ChEMBL database (version 14). A user may select proteins and list their ligands interactively and then design an activity profile iteratively by adding more targets or anti-targets to the selection and adjusting protein-specific activity thresholds. The compounds that meet the desired profile are displayed within the application (with links to ChEMBL) and the full collection can be saved as an SD file.You may even modify the code provided and so link the application to your own in-house private data source.
%T ChEMBLSpace – a graphical explorer of the chemogenomic space covered by the ChEMBL database %A N. Fechner %A G. Papadatos %A D. Evans %A J.R. Morphy %A S.C. Brewerton %A D. Thorner %A M. Bodkin %J Bioinformatics %D 2012 %O http://dx.doi.org/10.1093/bioinformatics/bts711
George -
Paper: Structural basis of ligand recognition in 5-HT3 receptors

The power and insight provided by structural biology into pharmacology can never be underestimated, and significant progress is now possible for previously challenging target systems for structure determination, most notably the GPCRs, which we regularly cover here on the ChEMBL-og. However, there are an increasing number of ion channel and transporter structures being determined. We are probably in sight of the time when the majority of pharmacology for known drugs can be placed in a three-dimensional structure setting. We live in exciting times!
Here is a paper on a further important drug target family - the ligand-gated ion channels - more specifically structural studies on the multimeric ligand-binding extracellular domain. The work involved the engineering of a more tractable homolog (Aplysia AChBP - Aplysia is a sea hare if you are interested) of the human 5HT3 receptor (5HT3R) to become more 5HT3-like in it's ligand binding properties (Pfam: Neur_chan_LBD PF02931). Complexes of this functional surrogate of 5HT3R with serotonin - the natural agonist ligand, and the drug antagonist granisetron were then determined and analysed. This structural surrogate approach has been tried several times previously (e.g. for nicotinic receptor ligands here).
There's some interesting mutagenesis data reported some of the tables, reporting the binding affinities for some of the mutants - this would be great training/validation data for some proteochemometrics studies, and has also got me wondering if there could be some useful general approximate predictive models for ligand-binding differences developed for cSNPs on the back of this sort of data - similar work for presumed protein stability has previously been developed (e.g. here)....
%A D. Kesters %A A.J. Thompson %A M. Brams %A R. van Elk %A R. Spurny %A M. Geitmann %A J.M. Villalgordo %A A. Guskov %A D.U. Helena Danielson %A S.C.R. Lummis %A A.B. Smit %A C. Ulens %T Structural basis of ligand recognition in 5-HT3 receptors %J EMBO Rep %D 2013 %V 14 %P 49-56 %O http://dx.doi.org/10.1038/embor.2012.18
jpo -
New Drug Approvals 2012 - Pt. XXVIII - Teduglutide (Gattex®)

ATC code: A16AX08Wikipedia: Teduglutide
On December 27, FDA approved Teduglutide (Tradename: Gattex®, Tradename: Revestive, Research Code: ALX-600, CAS# 197922-42-2), an analog of Glucagon-like peptide 2 (GLP2) expressed in Escherichia coli by recombinant DNA (rDNA) technology indicated for the treatment of adult patients with Short Bowel Syndrome (SBS).
Short Bowel Syndrome (MeSH: D012778) is a condition in which nutrients are not properly absorbed (malabsorption disorder) caused by surgical removal of small intestine, or rarely due to the complete dysfunction of large segment of bowel. More information about SBS can be found in Medscape.
Teduglutide is an analog of naturally occurring Glucagon-like peptide 2 (GLP2), which is 33 amino acid peptide created by specific post-translational proteolytic cleavage of proglucagon in endocrine L cells of the distal intestine. GLP2 is known to increase intestinal and portal blood flow, and inhibit gastric acid secretion.
Teduglutide binds to Glucagon-like peptide-2 receptors (GLP-2R) located in subpopulations of enteroendocrine cells of intestine, sub-epithelial myofibroblasts and enteric neurons of the submucosal and myenteric plexus. Activation of these receptors results in local release of multiple mediators including Insulin-like growth factor (IGF-1), nitric oxide (NO) and Keratinocyte growth factor (KGF).
Glucagon-like peptide-2 receptors (GLP-2R, CHEMBL5844, O95838), 553 amino acid long, is a receptor for glucagon-like peptide-2. The activity of this receptor is mediated by G proteins which activate adenylyl cyclase. The amino acid sequence of GLP-2R (human) is :
>GLP2R
MKLGSSRAGPGRGSAGLLPGVHELPMGIPAPWGTSPLSFHRKCSLWAPGRPFLTLVLLVS
IKQVTGSLLEETTRKWAQYKQACLRDLLKEPSGIFCNGTFDQYVCWPHSSPGNVSVPCPS
YLPWWSEESSGRAYRHCLAQGTWQTIENATDIWQDDSECSENHSFKQNVDRYALLSTLQL
MYTVGYSFSLISLFLALTLLLFLRKLHCTRNYIHMNLFASFILRTLAVLVKDVVFYNSYS
KRPDNENGWMSYLSEMSTSCRSVQVLLHYFVGANYLWLLVEGLYLHTLLEPTVLPERRLW
PRYLLLGWAFPVLFVVPWGFARAHLENTGCWTTNGNKKIWWIIRGPMMLCVTVNFFIFLK
ILKLLISKLKAHQMCFRDYKYRLAKSTLVLIPLLGVHEILFSFITDDQVEGFAKLIRLFI
QLTLSSFHGFLVALQYGFANGEVKAELRKYWVRFLLARHSGCRACVLGKDFRFLGKCPKK
LSEGDGAEKLRKLQPSLNSGRLLHLAMRGLGELGAQPQQDHARWPRGSSLSECSEGDVTM
ANTMEEILEESEI
Teduglutide, just like GLP2 and is 33 amino acids long, with a molecular weight of 3752.13 daltons. The recommended daily dose is 0.05 mg/kg body weight administered by subcutaneous injection once daily. The chemical structure was obtained from Chemblink.
Amino acid sequence of Teduglutide is : His-Gly-Asp-Gly-Ser-Phe-Ser-Asp-Glu-Met-Asn-Thr-Ile-Leu-Asp-Asn-Leu-Ala-Ala-Arg-Asp-Phe-Ile-Asn-Trp-Leu-Ile-Gln-Thr-Lys-Ile-Thr-Asp

After subcutaneous administration of Teduglutide in healthy individuals, the absolute bioavailability was 88% and maximum plasma concentrations was achieved in 3 to 5 hours, volume of distribution was 103 mL/kg and plasma clearance was approximately 123 mL/hr/kg with mean terminal half_life of approximately 2 hrs. Metabolic pathway of Teduglutide was not investigated in humans, however it is expected to be degraded into small peptides and amino acids via catabolic pathways similar to endogenous GLP-2.
The license holder is NPS Pharmaceuticals, and the product website is www.gattex.com.
Full prescribing information can be found here.
Ramesh
-
New Drug Approvals 2012 - Pt. XXIX - Pasireotide diaspartate (SIGNIFOR®)
 ATC code: H01CB05Wikipedia: PasreotideOn December 14, the FDA approved Pasireotide diaspartate (SIGNIFOR®, Research CodeL SOM-230, CAS# 396091-73-9, Pub-Chem : CID 9941444, KEGG : D10147), a cyclohexide with pharmacologic properties mimicking those of natural hormone Somatostatin for treatment of adult patients with Cushing's disease (CD) for whom pituitary surgery is not an option or has not been curative.Cushing's disease (CD) is a cause of Cushing's syndrome characterised by increased secretion of ACTH (adrenocorticotropic hormone) from the anterior pituitary. CD is a rare hormone disorder, and recent statistics indicate that the annual incidence is somewhere between 1 and 10 per million and is 3 times more common in women than in men (from NEMDIS). This is most often as a result of pituitary adenoma or due to excess production of hypothalamus Corticotropin releasing hormone (CRH). More information can be found in Medscape.
ATC code: H01CB05Wikipedia: PasreotideOn December 14, the FDA approved Pasireotide diaspartate (SIGNIFOR®, Research CodeL SOM-230, CAS# 396091-73-9, Pub-Chem : CID 9941444, KEGG : D10147), a cyclohexide with pharmacologic properties mimicking those of natural hormone Somatostatin for treatment of adult patients with Cushing's disease (CD) for whom pituitary surgery is not an option or has not been curative.Cushing's disease (CD) is a cause of Cushing's syndrome characterised by increased secretion of ACTH (adrenocorticotropic hormone) from the anterior pituitary. CD is a rare hormone disorder, and recent statistics indicate that the annual incidence is somewhere between 1 and 10 per million and is 3 times more common in women than in men (from NEMDIS). This is most often as a result of pituitary adenoma or due to excess production of hypothalamus Corticotropin releasing hormone (CRH). More information can be found in Medscape. The therapeutic activity of Pasireotide diaspartate is through binding to Somatostatin receptors (SSTRs). They belong to GPCR class of targets and there are five known SSTRs in human: SSTR1 (CHEMBL1917; P30872), SSTR2 (CHEMBL1804; P30874), SSTR3 (CHEMBL2028; P32745), SSTR4 (CHEMBL1853; P31391) and SSTR5 (CHEMBL1792; P35346). These receptor subtypes are expressed in different tissues under normal physiological condition. Corticotroph tumor cells from CD patients frequently over express SSTR5, whereas the other receptor subtypes are often not expressed or expressed at lower levels. Pasireotide diaspartate has a 40-fold increased affinity to SSTR5 than other SSTR analogs binds and activates the receptors resulting in inhibition of ACTH secretion, which leads to decreased cortisol secretion.
The therapeutic activity of Pasireotide diaspartate is through binding to Somatostatin receptors (SSTRs). They belong to GPCR class of targets and there are five known SSTRs in human: SSTR1 (CHEMBL1917; P30872), SSTR2 (CHEMBL1804; P30874), SSTR3 (CHEMBL2028; P32745), SSTR4 (CHEMBL1853; P31391) and SSTR5 (CHEMBL1792; P35346). These receptor subtypes are expressed in different tissues under normal physiological condition. Corticotroph tumor cells from CD patients frequently over express SSTR5, whereas the other receptor subtypes are often not expressed or expressed at lower levels. Pasireotide diaspartate has a 40-fold increased affinity to SSTR5 than other SSTR analogs binds and activates the receptors resulting in inhibition of ACTH secretion, which leads to decreased cortisol secretion. Since the targets of this drug belong to same family of receptors (somatostatin receptors), multiple sequence alignment of human SSTR1, 2, 3, 4 and 5 receptors was done using T-coffee which is shown above. The protein sequences (fasta format) of human SSTR1, 2, 3, 4 and 5 can be downloaded from this link here. (courtesy UniProt)
Since the targets of this drug belong to same family of receptors (somatostatin receptors), multiple sequence alignment of human SSTR1, 2, 3, 4 and 5 receptors was done using T-coffee which is shown above. The protein sequences (fasta format) of human SSTR1, 2, 3, 4 and 5 can be downloaded from this link here. (courtesy UniProt) Standard InChI : 1/C58H66N10O9/c59-27-13-12-22-46-52(69)64-47(30-38-23-25-42(26-24-38)76-36-39-16-6-2-7-17-39)53(70)66-49(31-37-14-4-1-5-15-37)57(74)68-35-43(77-58(75)61-29-28-60)33-50(68)55(72)67-51(40-18-8-3-9-19-40)56(73)65-48(54(71)63-46)32-41-34-62-45-21-11-10-20-44(41)45/h1-11,14-21,23-26,34,43,46-51,62H,12-13,22,27-33,35-36,59-60H2,(H,61,75)(H,63,71)(H,64,69)(H,65,73)(H,66,70)(H,67,72)/t43-,46+,47+,48-,49+,50+,51+/m1/s1/f/h61,63-67HSmiles : NCCCC[C@H]1C(N[C@H](C(N[C@H](C(N2[C@H](C(N[C@H](C(N[C@@H](C(N1)=O)CC1=CNC3=CC=CC=C13)=O)C1=CC=CC=C1)=O)C[C@H](C2)OC(NCCN)=O)=O)CC2=CC=CC=C2)=O)CC2=CC=C(C=C2)OCC2=CC=CC=C2)=OIupac Name : (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3-ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18-hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt
Standard InChI : 1/C58H66N10O9/c59-27-13-12-22-46-52(69)64-47(30-38-23-25-42(26-24-38)76-36-39-16-6-2-7-17-39)53(70)66-49(31-37-14-4-1-5-15-37)57(74)68-35-43(77-58(75)61-29-28-60)33-50(68)55(72)67-51(40-18-8-3-9-19-40)56(73)65-48(54(71)63-46)32-41-34-62-45-21-11-10-20-44(41)45/h1-11,14-21,23-26,34,43,46-51,62H,12-13,22,27-33,35-36,59-60H2,(H,61,75)(H,63,71)(H,64,69)(H,65,73)(H,66,70)(H,67,72)/t43-,46+,47+,48-,49+,50+,51+/m1/s1/f/h61,63-67HSmiles : NCCCC[C@H]1C(N[C@H](C(N[C@H](C(N2[C@H](C(N[C@H](C(N[C@@H](C(N1)=O)CC1=CNC3=CC=CC=C13)=O)C1=CC=CC=C1)=O)C[C@H](C2)OC(NCCN)=O)=O)CC2=CC=CC=C2)=O)CC2=CC=C(C=C2)OCC2=CC=CC=C2)=OIupac Name : (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3-ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18-hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt
Pasireotide diaspartate is a Somatostatin analog and is cyclohexapeptide with pharmacologic properties mimicking those of the natural hormone somatostatin. The recommended dosage range of Pasireotide diaspartate is 0.3 to 0.9 mg administered as subcutaneous injection twice a day.Pasireotide diaspartate demonstrates approximately linear pharmacokinetics for a dose range from 0.0025 to 1.5 mg in healthy patients with dose proportional Cmax and AUC and Tmax of 0.25 to 0.5 hrs. Apparent volume of distribution (Vz/F) was >100 L with plasma concentration of about 91%. Plasma protein binding was moderate (88%). It is shown to be metabolically stable in human liver and kidney and is eliminated mainly via hepatic clearance.Full prescribing information can be found here.The license holder is Novartis Pharmaceuticals, and the product website is www.signifor.com. -
New Drug Approvals 2012 - Pt. XXX - Crofelemer (Fulyzaq TM)

ATC code: not yet assignedWikipedia: CrofelemerOn December 31 2012, the FDA approved Crofelemer for the treatment of diarrhea caused by antiretroviral medication regimens taken by patients with HIV/AIDS.
Diarrhea is a frequent adverse effect of antiretroviral medication - however, it can also be caused by secondary infections of the gastrointestinal tract or the virus itself. The loss of fluids incurred by diarrhea can lead to dehydration and electrolyte imbalance. As a side-effect of antiretroviral medication it also reduces patient compliance with a prescribed medication regimen.
Crofelemer alleviates the symptoms of HIV-associated diarrhea by limiting the amount of chloride ions that are pumped into the intestinal lumen, thus also retaining sodium ions and water. Crofelemer is a natural product oligomer that is not orally bioavailable but acts locally on the intestinal surface. It was shown to inhibit two distinct intestinal chloride channels expressed in the luminal membrane of gut epithelial cells. One of them, the cystic fibrosis transmembrane conductance regulator (CFTR, Uniprot: P13569, CHEMBL4051) is a member of the ABS transporter family, the other is a calcium-activated chloride channel (CaCC) and is called Anoctamin-1 (TMEM16A, Uniprot: Q5XXA6). Anoctamin-1 is currently the only known CaCC expressed at the luminal membrane of gut epithelial cells but crofelemer might also inhibit other CaCCs.

Crofelemer is extracted from the red latex of the South American tree Croton lechleri and is an amorphous red-brown powder. The oligomer is a random sequence of (+)-catechin, (-)-epicatechin, (+)gallocatechin and (-)-epigallocatechin monomers (on average 5 to7.5) and average mass of 1500Da to 2300Da.
Crofelemer is dosed twice-daily in delayed release tablets containing 125mg of the active ingredient. It is practically not absorbed into the blood and typical pharmacokinetic parameters are thus not applicable.
Crofelemer was initially developed by Shaman Pharmaceuticals and then continued by Napo Pharmaceuticals (Forbes has a short article on Crofelemer's development). At the moment, Salix Pharmaceuticals holds a license for marketing in Japan, North America and Europe while Glenmark Pharmaceuticals has exclusive rights to marketing in 140 developing countries including India (but excluding China).
The commercial name for crofelemer is Fulyzaq.
-
New Drug Approvals 2012 - Pt. XXVI - cabozantinib (COMETRIQTM)

ATC code: not yet assigned Wikipedia: Cabozantinib
On November 29th, the FDA approved CometriqTM(cabozantinib, research code XL-184) for the treatment of patients with progressive, metastatic, medullary thyroid cancer (MTC). MTC is a rare type of thyroid cancer accounting for 5-10% of all diagnosed thyroid cancers (CRUK). A quarter of MTC cases are familial and are caused by a mutation in the RET gene. Clinical studies showed that cabozantinib increases progression-free survival time to 11.2 months in comparison with 4 months for placebo.
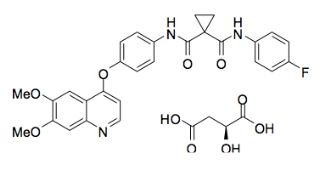
Cabozantinib (research codes: XL-184, BMS-907351; IUPAC: Cabozantinib (S)-malate is described chemically as N-(4-(6,7-dimethoxyquinolin-4-yloxy)phenyl)-N’-(4-fluorophenyl)cyclopropane- 1,1-dicarboxamide, Standard InChI: ONIQOQHATWINJY-UHFFFAOYSA-N) is dosed as the (S)-malate salt. The parent molecule, cabozantinib, has a molecular weight of 501.5 and a ACD/LogP of 3.08. The effective half-life is ~55 hours, the oral volume of distribution (V/F) is ~349 L, and the clearance (CL/F) at steady-state is estimated to be 4.4 L/hr.
Cabozantinib has been given a boxed warning due to sever, sometimes fatal haemorrhaging in 3% of patients and gastrointestinal perforations in 3% and fistula formation in 1% of treated patients.
Cabozantinib is a multi-kinase inhibitor, and inhibits the following tyrosine kinases in vitro: RET (P07949), MET (P08581), FLT1 (aka VEGFR1, P17948), KDR (aka VEGFR2, P35968), FLT4 (aka VEGFR3, P35916), KIT (P10721), NTRK2 (TRKB Q16620), FLT3 (P36888), AXL (P30530), and TEK (aka TIE2, Q02763).
Prescribing information can be found here.
The license holder for CometriqTM is Exelexis Inc and the product website can be found here -
I can't get no statin-faction

I've just got to 48 years old (or thirty-eighteen, in my newly favoured numbering scheme) and am taking stock of my personal health and underlying disease risks (I've just spat into my 23andMe tube - and I've got a the offer of guided expert interpretation of the results - I'm really lucky to work at the EBI for this sort of stuff). I already take low-dose aspirin, for it's reported protective effects in DVT and for various cancers. My kids say that it's about 38 years too late to start worrying about my health. But I feel frail suddenly.
Statins are great drugs, they significantly reduce the risk of cardiovascular events (that's a bit of a euphemism, isn't it?), and also there is a lot of evidence emerging for statins being potentially protective in a whole bunch of other inflammatory and proliferative disorders. Google is you friend in finding these sort of reports, so I won't evidence them here.
Simvastatin, a drug with huge patient exposure and safety data is now available in the UK (and several other places) Over The Counter (OTC) in a low dose form (10 mg) - the risk reduction in major CV events is 10% after a years dosing, and a third over three years at this dose (see here for a simple Q&A document) - this is pretty profound in my view. In fact there are many people who believe that above a threshold age, that most people should take statins.
An issue though is cost, if there was greatly increased coverage of people receiving statins, then the cost to the NHS (and the burden on the taxpayer) would be huge (remember this is a UK view, your local situation may well be different). With a well tolerated and safer form of statins (i.e. low dose), and general encouragement of the health benefits, and with them being available OTC (where the customer pays directly for the drug with no State involvement), at a population level, the health benefits would be great, and the cost of the benefit would be borne by the patient. Great all round! Of course, people at significantly higher than background risk can still get a prescription, monitoring and support, a higher dose form for more aggressive reduction of cholesterol levels).
So as a reasonably informed consumer, I decided to add statins to my daily routine. So I visited two large local pharmacies, only to be told that they no longer stock the OTC form (but there were visible packs of POM statins in all the pharmacies I visited. After wasting some time, I asked why they didn't stock them and was told the supplier doesn't deliver them any more, for both pharmacy chains. I called the helpline of Boots Company plc - and spoke to a representative, and they could not find OTC statins on their product list - quite odd - so I then asked the question - do they have any stores that actually stocked 10 mg OTC simvastatin? They couldn't answer, but put me through to a pharmacist at a major local branch - they were helpful, but confirmed OTC Simvastatin was an order only item. Hardly the best way to get 1) major population scale CV health benefits and 2) a shift in cost from State to patient.
It made me think a little - why is the supply chain not supplying OTC statins; but obviously, looking at the shelves in the pharmacies, having no problem with getting Branded Statins out?
Anyway, I am determined to get my OTC statins at my cost, and I'll post how I get on!
jpo
PS I also went to the dentist today and got two big fillings, and all I can say to the scientists who discovered and developed lidocaine is - you are Gods! Seriously, it was the first time I've ever opted for pain relief during dentistry, and I cannot believe what a difference it made, or how stupid I have been in the past.







