-
ChEMBL 14 Released

We are pleased to announce the release of ChEMBL_14. This latest version of the ChEMBL database contains:
- 1,384,479 compound records
- 1,213,242 distinct compounds
- 644,734 assays
- 10,129,256 bioactivities
- 9,003 targets
- 46,133 documents
- 10 data sources
- DrugMatrix - in vitro pharmacology assays for 870 therapeutic, industrial and environmental chemicals against 132 protein targets.
- GSK Published Kinase Inhibitor Set - two data sets screening this compound library have been deposited by Nanosyn and the University of North Carolina.
You download the ChEMBL_14 data from our ftpsite, but please refer to the chembl_14 release notes for a full list updates, changes and also details on planned schema changes in forthcoming ChEMBL releases.
-
ChEMBL delivers a new malaria data service
 As announced also here, a new malaria data service is available today to researchers around the globe sponsored by the Medicines for Malaria Venture (MMV).
As announced also here, a new malaria data service is available today to researchers around the globe sponsored by the Medicines for Malaria Venture (MMV).
The service provides access to hundreds of thousands of data points on malaria-related compounds, assays and targets, thus facilitating research for this neglected tropical disease. Inspired by the successful ChEMBL interface, a user may query the database using keywords, synonyms, chemical structures or protein sequences, review and filter the hits using tables or charts and then download the resulting subset. Data provenance is also provided so that the user can filter on the data sources they are interested in, such as scientific literature, the GlaxoSmithKline TCAMS, Novartis GNF and St Jude datasets or the MMV open access Malaria Box.
Based on the ChEMBL update cycle, the malaria data database will be regularly updated with depositions by academic and industrial groups who wish to share their malaria screening data with the rest of the research community. More depositions are currently being processed and will be available shortly. -
Drug and Targets
 ‘HIV reverse transcriptase is the target of aciclovir’ – easy to say and it’s sort of correct - it’s the sort of statement that in the vernacular of drug discovery, most people would accept without the blink of an eye. This sentence strikes at the core of the concept of a target (HIV reverse transcriptase) for a drug (aciclovir). However, there is much detail under this simple statement that captures some of the complexities of the representation and storage of bioactivity data.
‘HIV reverse transcriptase is the target of aciclovir’ – easy to say and it’s sort of correct - it’s the sort of statement that in the vernacular of drug discovery, most people would accept without the blink of an eye. This sentence strikes at the core of the concept of a target (HIV reverse transcriptase) for a drug (aciclovir). However, there is much detail under this simple statement that captures some of the complexities of the representation and storage of bioactivity data. Aciclovir is an inhibitor of HIV replication, so it is targeted to the virus itself – and indeed this can be a useful way of thinking about the mechanism and effect of aciclovir (and all other antiretroviral drugs). We know a lot about HIV-replication and infection, of which the reverse transcriptase function is an essential part, shared across all retroviruses, and is the process that aciclovir blocks. Due to the intense research on this devastating pathogen, we know a lot of detail about HIV (there was a striking paper on Nature on this a few years ago) and this ‘systems-level’ information can also be represented in terms of a network/pathway, in a resource like Reactome. Being able to tag this pathway with a drug is a useful thing to do as well – but we are typically interested in the more molecular and biochemical aspects of how a drug works – the molecular basis of it’s action.Firstly – HIV is a name of a family of viruses, HIV-1 and HIV-2 being the major forms, each of these can be further classified into subtypes/strains, e.g. HIV-1A, and within each of these strains, it’s appropriate to think of an infected person as containing a constantly changing ensemble of sub-strains. The entire family is related in sequence, but the key point is that the sequences differ - between HIV-1 and HIV-2 the differences are relatively substantial, and between the particular pool of viruses within a patient they are typically minor differences. So how should you store the organism/target (and an associated particular sequence) for this case?A comfort here is that it, in most cases, doesn’t really matter – any sequence of a native HIV-1 virus is basically OK, since aciclovir will probably usefully inhibit these, and the affinity/potency differences will be negligible. In fact, aciclovir is active as an inhibitor against both the HIV-1 and HIV-2 viruses. A big, big exception though is for strains of virus that have been under selective pressure following treatment with aciclovir – clinically resistant sequences are rapidly selected, and here the most frequent sequence in an infected individual will have significantly lower binding affinity for aciclovir. Usually these differences are near the drug-binding site, but not always. So for cases like this, it makes sense to try and store the sequence of the resistant strain – but of course each drug will have it’s own ensemble of resistant strains, and so it becomes complex. However, in order to understand selectivity profiles and risks, the management of these differences is crucial – as it is for intra-human sequence variation.So, HIV-1 is a virus, it has a genome, and some sequences, there are a number of genes within HIV-1 – XXXX of them, and the major ones are env, gag, and pol (there are also a bunch of others including, tat, rev, vpr, vif, nef, vpu and tev). These genes were named after the envelope, group-specific antigen, and polymerase functions early in the study of HIV-1. It turns out that the reverse transcriptase (RT) is part of the pol gene, and the pol gene also encodes the integrase and proteinase (both also the ‘targets’ of clinically successful drugs). The key word here is ‘part of’.RT is part of the pol gene – it requires cleavage from the precursor polyprotein to become catalytically active (and to be inhibited by aciclovir). The cleavage from the polyprotein is performed by a specific proteinase encoded in the HIV-1 genome (called PR) – this proteolytic activity is essential, and there are a class of drugs targeted against HIV-1 PR. So the gene sequence itself doesn’t contain all the information to capture the functional activity of RT – you need to know the sequence of the mature protein.It’s a little bit more complex than that though – the functional RT is actually an obligate dimer of two RT sequences – and a little more complicated than that yet, it isn’t a homo-dimer (two identical chains) but a heterodimer made up of two different length chains called p81 and p73 (the numbers refer to the approximate sizes of the proteins from early gel experiments).So, we’re getting there, slowly. ‘The p51/p66 RT heterodimer of HIV-1A is the target of aciclovir’ is better.Of course, in an ideal database, we’d need to be able to store this target information in a usable form, that can then be generalised to new systems. This isn’t just some nerdery, this detailed representation is essential for things like docking, understanding the consequences of mutations, etc.We know the 3-D structure of the mature dimeric form of HIV-1 RT and it is in fact composed of a series of distinct structural domains, and ligand binding is often associated with binding to a specific domain within these multidomain sequences. So storing the ligand binding domain(s) is a useful thing too, if you want to be able to generalise the observations across new data.Enough of the target for now!Now, let's think about the drug for a moment – aciclovir – an old drug, rescued from it’s original application as a potential anti-cancer to an anti-viral. Is aciclovir an inhibitor of this functional heterodimer?No. It isn’t.What is an inhibitor though is an active metabolite of aciclovir – specifically the triphosphate form. Aciclovir is an example of a prodrug – inactive (against it’s efficacy target) in the dosed form, and requiring specific metabolic events to occur before it is active against it’s target.
Aciclovir is an inhibitor of HIV replication, so it is targeted to the virus itself – and indeed this can be a useful way of thinking about the mechanism and effect of aciclovir (and all other antiretroviral drugs). We know a lot about HIV-replication and infection, of which the reverse transcriptase function is an essential part, shared across all retroviruses, and is the process that aciclovir blocks. Due to the intense research on this devastating pathogen, we know a lot of detail about HIV (there was a striking paper on Nature on this a few years ago) and this ‘systems-level’ information can also be represented in terms of a network/pathway, in a resource like Reactome. Being able to tag this pathway with a drug is a useful thing to do as well – but we are typically interested in the more molecular and biochemical aspects of how a drug works – the molecular basis of it’s action.Firstly – HIV is a name of a family of viruses, HIV-1 and HIV-2 being the major forms, each of these can be further classified into subtypes/strains, e.g. HIV-1A, and within each of these strains, it’s appropriate to think of an infected person as containing a constantly changing ensemble of sub-strains. The entire family is related in sequence, but the key point is that the sequences differ - between HIV-1 and HIV-2 the differences are relatively substantial, and between the particular pool of viruses within a patient they are typically minor differences. So how should you store the organism/target (and an associated particular sequence) for this case?A comfort here is that it, in most cases, doesn’t really matter – any sequence of a native HIV-1 virus is basically OK, since aciclovir will probably usefully inhibit these, and the affinity/potency differences will be negligible. In fact, aciclovir is active as an inhibitor against both the HIV-1 and HIV-2 viruses. A big, big exception though is for strains of virus that have been under selective pressure following treatment with aciclovir – clinically resistant sequences are rapidly selected, and here the most frequent sequence in an infected individual will have significantly lower binding affinity for aciclovir. Usually these differences are near the drug-binding site, but not always. So for cases like this, it makes sense to try and store the sequence of the resistant strain – but of course each drug will have it’s own ensemble of resistant strains, and so it becomes complex. However, in order to understand selectivity profiles and risks, the management of these differences is crucial – as it is for intra-human sequence variation.So, HIV-1 is a virus, it has a genome, and some sequences, there are a number of genes within HIV-1 – XXXX of them, and the major ones are env, gag, and pol (there are also a bunch of others including, tat, rev, vpr, vif, nef, vpu and tev). These genes were named after the envelope, group-specific antigen, and polymerase functions early in the study of HIV-1. It turns out that the reverse transcriptase (RT) is part of the pol gene, and the pol gene also encodes the integrase and proteinase (both also the ‘targets’ of clinically successful drugs). The key word here is ‘part of’.RT is part of the pol gene – it requires cleavage from the precursor polyprotein to become catalytically active (and to be inhibited by aciclovir). The cleavage from the polyprotein is performed by a specific proteinase encoded in the HIV-1 genome (called PR) – this proteolytic activity is essential, and there are a class of drugs targeted against HIV-1 PR. So the gene sequence itself doesn’t contain all the information to capture the functional activity of RT – you need to know the sequence of the mature protein.It’s a little bit more complex than that though – the functional RT is actually an obligate dimer of two RT sequences – and a little more complicated than that yet, it isn’t a homo-dimer (two identical chains) but a heterodimer made up of two different length chains called p81 and p73 (the numbers refer to the approximate sizes of the proteins from early gel experiments).So, we’re getting there, slowly. ‘The p51/p66 RT heterodimer of HIV-1A is the target of aciclovir’ is better.Of course, in an ideal database, we’d need to be able to store this target information in a usable form, that can then be generalised to new systems. This isn’t just some nerdery, this detailed representation is essential for things like docking, understanding the consequences of mutations, etc.We know the 3-D structure of the mature dimeric form of HIV-1 RT and it is in fact composed of a series of distinct structural domains, and ligand binding is often associated with binding to a specific domain within these multidomain sequences. So storing the ligand binding domain(s) is a useful thing too, if you want to be able to generalise the observations across new data.Enough of the target for now!Now, let's think about the drug for a moment – aciclovir – an old drug, rescued from it’s original application as a potential anti-cancer to an anti-viral. Is aciclovir an inhibitor of this functional heterodimer?No. It isn’t.What is an inhibitor though is an active metabolite of aciclovir – specifically the triphosphate form. Aciclovir is an example of a prodrug – inactive (against it’s efficacy target) in the dosed form, and requiring specific metabolic events to occur before it is active against it’s target. ‘The p51/p66 RT heterodimer of HIV-1A is the target of active metabolite of aciclovir’ is getting there.More nerdery you cry – well no. If you wanted to discover computationally that aciclovir was useful as a drug for HIV – you’d need to know (or store) the active triphosphate form (there are also come intermediate forms on the way to the triphosphate that should probably be considered too). Of course, the body also ‘sees’ the originally dosed aciclovir, so you may want to store that to, dock it to host proteins for side effects, etc.At this stage we’ve probably got a detailed enough representation of the drug-target complex to allow us to do some reliable and useful things with the data.It is worth going to a higher level of detail though, since it illustrates another important point.Aciclovir triphosphate binds in a specific binding site of HIV-1 RT, at the catalytic site – this is definitively known from enzymatic and structural studies, since aciclovir is a nucleoside analogue, this site is known as the nucleoside site. Sequence changes around this nucleoside site can rapidly be selected for to give rise to resistant variants. Knowing where the drug aciclovir binds can aid both sequence/resistance analysis studies, 3-D modelling, and also help in docking experiments, since it’s possible to focus studies on a known functional site.There’s a second class of drug, NNRTIs – non-nucleoside reverse transcriptase inhibitors. Prototypical of these is efiravenz. These are very different in chemical structure to nucleoside analogues, and in fact bind at a different site – an ‘allosteric’ site, that isn’t formed until the ligand binds. Resistance can a does arise for this class of inhibitor too, but because the drug binds at a different site, a different constellation of residues is involved in resistance. Interestingly, this site doesn’t exist at all in the closely related HIV-2 enzyme, and so NNRTIs are essentially inactive against HIV-2.So this site is allosteric – what does this mean – well since the structure of the protein varies during ligand binding – it is important to keep track of these different possible conformational states – essential if one wants to do docking, etc. At the tip of this target taxonomy we have to think about a particular conformational substrate of a protein.So there are two target sites in HIV RT, the nucleoside and the NNRTI site, so perhaps we should state….‘The nucleoside binding site of p51/p66 RT heterodimer of HIV-1A is the target of active metabolite of aciclovir’
‘The p51/p66 RT heterodimer of HIV-1A is the target of active metabolite of aciclovir’ is getting there.More nerdery you cry – well no. If you wanted to discover computationally that aciclovir was useful as a drug for HIV – you’d need to know (or store) the active triphosphate form (there are also come intermediate forms on the way to the triphosphate that should probably be considered too). Of course, the body also ‘sees’ the originally dosed aciclovir, so you may want to store that to, dock it to host proteins for side effects, etc.At this stage we’ve probably got a detailed enough representation of the drug-target complex to allow us to do some reliable and useful things with the data.It is worth going to a higher level of detail though, since it illustrates another important point.Aciclovir triphosphate binds in a specific binding site of HIV-1 RT, at the catalytic site – this is definitively known from enzymatic and structural studies, since aciclovir is a nucleoside analogue, this site is known as the nucleoside site. Sequence changes around this nucleoside site can rapidly be selected for to give rise to resistant variants. Knowing where the drug aciclovir binds can aid both sequence/resistance analysis studies, 3-D modelling, and also help in docking experiments, since it’s possible to focus studies on a known functional site.There’s a second class of drug, NNRTIs – non-nucleoside reverse transcriptase inhibitors. Prototypical of these is efiravenz. These are very different in chemical structure to nucleoside analogues, and in fact bind at a different site – an ‘allosteric’ site, that isn’t formed until the ligand binds. Resistance can a does arise for this class of inhibitor too, but because the drug binds at a different site, a different constellation of residues is involved in resistance. Interestingly, this site doesn’t exist at all in the closely related HIV-2 enzyme, and so NNRTIs are essentially inactive against HIV-2.So this site is allosteric – what does this mean – well since the structure of the protein varies during ligand binding – it is important to keep track of these different possible conformational states – essential if one wants to do docking, etc. At the tip of this target taxonomy we have to think about a particular conformational substrate of a protein.So there are two target sites in HIV RT, the nucleoside and the NNRTI site, so perhaps we should state….‘The nucleoside binding site of p51/p66 RT heterodimer of HIV-1A is the target of active metabolite of aciclovir’
Another away to think about this is as a hierarchy
Aciclovir triphosphate is a....
- Retrovirus replication inhibitor
- HIV replication inhibitor
- HIV-1A replication inhibitor
- Reverse Transcriptase Inhibitor
- p55/p61 RT inhibitor
- nucleoside site binder
Imagine for a second that ‘HIV-3’ is sequenced, and we need a new drug quickly – we can sequence the genome pretty quickly and cheaply nowadays, but hopefully the complexity above will show that the transformation from the gene sequence to a useful object to be analysed as a target is a complex one, requiring a lot of tacit knowledge of the particular system.Don’t worry, not everything is as complicated as this example, and it is one of my favourites, since there are so many twists and turns in this particular case. But you must, you simply must, now be wondering how we currently do, and in the future will, cope with this sort of thing in ChEMBL. Well – that will be the subject of a future post!If there’s interest, I can add references and some background links to this post – let me know if you’d be interested in the comments. -
Carbon and Nitrogen and Oxygen
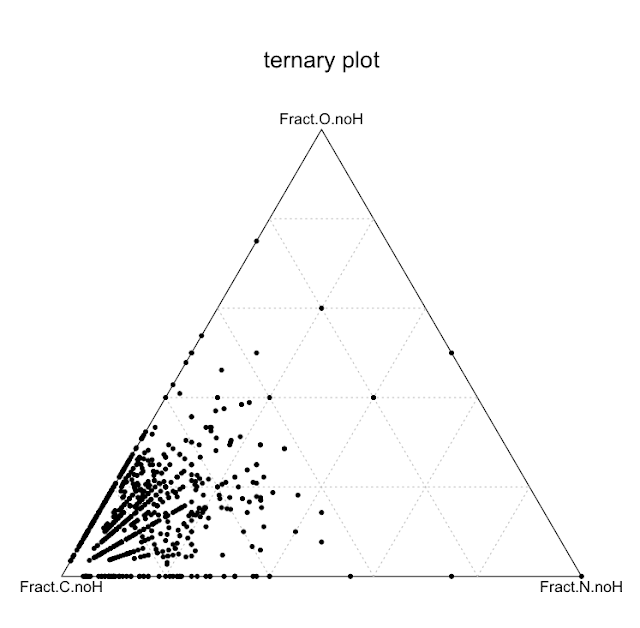
There was a post a few weeks ago about elemental compositions - simple cases of Carbon oxides, nitrogen oxides, etc. Well, I've processed ChEMBL to do the analysis across all of the compounds - very simple analysis - select all formulas of all compounds, select only those that are contain only elements restricted to the set CHNO, and then plot the heavy element (i.e. C N or O) fractions on a ternary plot.
Here it is (click image for larger).
So, the compounds that chemists make are mostly carbon-based, and of course, there's no contours on this version of the plot - hardly a big surprise, that's why they are organic, but things get more interesting when you think about using these data as filters/heuristics for expectation values for the sort of things that chemists could make, for spotting unusual compounds, etc. etc. More later on this...
Update: Here is the plot for drugs. As you will see there are some differences...
-
Paper - Combinatorial Drug Therapy For Cancer In The Post-genomic Era
 There's a great paper just out from some of our collaborators - Bissan Al-Lazikani, Udai Banerji and Paul Workman, of the ICR. It reviews drug combination strategies for cancer, and some of the molecular features of effective drug combinations.
There's a great paper just out from some of our collaborators - Bissan Al-Lazikani, Udai Banerji and Paul Workman, of the ICR. It reviews drug combination strategies for cancer, and some of the molecular features of effective drug combinations.
A link to the paper is here.
%A B. Al-Lazikani %A U. Banerji %A P. Workman %T Cobminatorial Drug Therapy For Cancer in the Post-genomic Era %J Nat. Biotech. %V 30 %P 1-13 %D 2012 %O DOI:
-
Compound Clean Up and Mapping (Posted by Louisa)

This new blog post has been created due to popular demand and user requests. I hope that this is useful for you.
After being manually extracted from the primary literature, a compound can be only loaded into the ChEMBL database after it has been run through our in-house clean-up protocol. This protocol utilises Accelrys's Pipeline Pilot software and has evolved a lot over the past three years. The clean-up protocol is used to prevent any structures from being loaded that could be incorrect, not properly charged or contain bad valences. We also use it to map the structures to already existing compounds in ChEMBL.
Historically, the clean-up protocol was very simple with just a few components to squeeze out any unwanted structures. Initially, we were mostly concerned about having uneven charges (e.g. charged counter ion but neutral parent) or quaternary nitrogen-containing compounds without a counter ion at all. Over the past 14 releases, the clean-up has become more sophisticated and now takes into account steroid backbone stereochemistry, inorganic salts and bad valences, amongst other things. A lot of the additions and adjustments have come from stumbling across little subsets of compounds that we hadn't thought of looking for before, and then warranted a consistent cleanup. This work has also led to the development of a series of business rules applied to consistently represent a functional group (for example, nitro groups).
Once the new compounds have been cleaned up, they need to be checked to see if they already exist in the database. Initially, this was done by mapping to the standard InChI, but it was soon realised that not all papers display the correct structure, if any structure at all, or it may not be extracted from the publication exactly as shown. This was causing duplicate compounds to be loaded into ChEMBL. Therefore, it was decided that a better initial mapping would be to use the extracted compound name and compare it to the many stored compound names in the database. This reduced a lot the duplicate entries. Once the new compounds have been mapped on the name, the remaining compounds are then mapped on their standard InChI. For those that don't match either a name or an InChI, I create a text file of their names and cast an eye over them to see if there are any that can still be mapped. I have been able to catch a couple of odd ones now and again via this last check, so it's definitely worth doing.
The compound clean-up is always a 'work in progress' and open to new suggestions for filtering out any compound or group of compounds that could do with further checking. If anyone would like to know more about the clean-up protocol or to send us some suggestions, please email chembl-help@ebi.ac.uk
-
Conference: New Horizons in Toxicity Prediction, September 2012, Cambridge, UK

A great looking conference on toxicity prediction to be held at Downing College, Cambridge on September 5th and 6th 2012 - a fantastic line up of speakers, and organised by one of our favourite collaborators LHASA Limited.
Details of the conference, registration, etc. are here. -
WT Course - Computational Approaches to Drug Discovery

So, the course is over for another year. It was really good fun to do, and thanks to all the attendees who made it really rewarding for us all. There is plenty for us to think about in what you asked, and we'll try and include a few more things in the ChEMBL interface. An especial thanks goes out to our visiting lecturers - John Irwin, Noel O'Boyle, Markus Sitzmann, Val Gillet, Andreas Bender, Darren Green, Bissan Al-Lazikani and Mike Barnes.
The picture is of (most of) the course attendees and the faculty on the last day, it was raining, much like every day that week, so an indoor photo. It looks cool.
Till next year!!






