-
Kinase inhibitor targets by highest phase

With a little bit of data wrangling I was able to convert the kinase clinical data we have to a form which shows the highest phase achieved for a particular target class - major distinct known activities were clustered together, and not surprisingly, the largest class was then classified as "multi" - i.e. significant known activity across multiple distinct branches of the kinome tree.
A little bit of explanation of the graph, blue = phase 1, green = phase 2, yellow = phase 3 and blue = launched. Targets are the x-axis, and the number of compounds that have reached that clinical phase are on the y-axis.
The graph is crappy - I couldn't get Numbers to behave - sorry, but c'est la vie. (Click image for larger)
If you want the data yourself in spreadsheet form mail me. -
Atlas CBS Server
We have set up a server for some of the ligand efficiency mapping work of Cele Abad-Zapatero and colleagues. The AtlasCBS server is at https://wwwdev.ebi.ac.uk/chembl/atlascbs/intro.jsp - note this is on our dev server, so if some of the links out are goofy, or look different, check the url!
The site and work is pretty well documented, so go on, have an explore - and any feedback you may have, leave a comment to this blog posting. -
Data Integration - The Ontogeny of Chemical Data
It is great to be able to perform confederated queries across data sources, really great. The greatness of this leads to the development of common representational and access standards (things like MIABE, InChI, and PSIQUIC, as examples that we are involved in ourselves). This ability to take data out of silos and share has arguably been one of the defining elements of science in the last ten years, and the generalisation of this data sharing and representation via semantic web technologies may become one of the defining features of the next decade. One of the issues though is that of data provenance, and primacy, and this is an issue that we have started to think about, and plan for in our own data integration efforts.
Data enters the 'system' somewhere in a 'database'; and these bundles are then 'licensed' to the community. The licenses may be silent, ambiguous, as clear-as-a-bell, shockingly unacceptable, or whatever - and there is a clear need for standardisation of licenses, or at the very least the requirement that license terms are freely available on the resource web site.
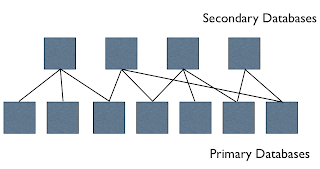
There are arguably two basic types of chemical databases at the moment - primary and secondary - primary databases are the first point of entry of that on the internet, or to the user community, and the groups responsible for these typically focus on data novelty (providing something that others don't) and also typically care about curation and indexing of the data. The current funding system does a pretty good job minimising overlap between funded primary databases. Primary data can be a one-off effort, a passive archive, or regularly updated with new content and curated; and there are many other nuances to consider. These primary databases tend to have a theme - spectroscopic data, or bioactivity data, or synthesis, etc., and be relatively small. Together they are the substrate for the secondary databases. There are arguably (again this is all just top of the head thinking) two types of integration that people typically do - 'vertical' and 'horizontal' - by 'vertical' I mean bundling all the chemical objects together, or bundling all the non-redundant proteins together, or bundling all protein structures and protein models together, and by 'horizontal' I mean integrating across different object classes, e.g. protein structures and small molecule ligands, or genes and protein structures, etc. The 'vertical secondary' databases typically add little new data, but confederate primary data content, reduce redundancy, provide cross-references, etc. but they often add significant value in terms of new descriptors, services, etc. It is also often a lot easier to curate data when integrated across other resources. Secondary databases may be physical (in that copies are made of the primary databases and all loaded together) or virtual (they query the primary resources through an API).
Secondary (vertical) databases do a great job of integration across these disparate resources, allowing queries across unlinked primary databases, and this is a required task for the more challenging horizontal integration. This horizontal data integration is probably where the majority of impact and insights from data are going to come from - for example there are great opportunities to integrate drug pharmacokinetic and pharmacodynamic data with human genetic variation data, to look for opportunities to deliver currently oral drugs topically, or to mine patent/marketing exclusivity expiries and look for arbitrage opportunities between diseases/healthcare systems, for the more entrepreneurial.
So some examples of primary and secondary chemistry databases - we like to think of ChEMBL as a primary database (more specifically the literature and deposited data, this makes up the majority of the data we have). We think of ChemSpider as a secondary database, and some databases like PubChem are arguably a mix of primary (for the NIH bioactivity data) and secondary (for compound catalogues and from other databases).
Is this view of chemical databases useful at all? Maybe not. But it maybe poses a couple of questions.
- What are the optimal mechanisms for curation and error correction of data? Is it performed at the secondary or primary levels?
- What are the primary and secondary resources - is it worth providing tracking of data provenance ? Should there be a standard format for the manifest of secondary resources?
- Secondary resources need to have effective update and correction capabilities driven by updates in their underlying primary resources (this is quite a poor area at the moment it seems - loaded once, a few years ago, doesn't mean that a secondary resource contains the best view of the primary data)?
- How is licensing addressed in secondary resources? - I've found quite a few examples of where I can't download a dataset from it's primary website, yet it is contained in a secondary resource, nominally freely available.
- Where should funding focus go - to standardising access and indexing of primary resources, or into consolidating data into secondary resources.
-
The EBI has joined the Open PHACTS project
 Just a quick note to say that we have joined the Open PHACTS Project, and as our local contribution (there are multiple strands to the EBI's involvement) we will be enhancing rdf/SPARQL access to ChEMBL, and also developing some improved semantic indexing of some of the assays and targets.
Just a quick note to say that we have joined the Open PHACTS Project, and as our local contribution (there are multiple strands to the EBI's involvement) we will be enhancing rdf/SPARQL access to ChEMBL, and also developing some improved semantic indexing of some of the assays and targets.
Details of the project itself are on the Open PHACTS website http://www.openphacts.org/. -
New Drug Approvals 2012 - Pt. IV - Vismodegib (ERIVEDGETM)
Wikipedia: Vismodegib
On Jan 31st 2012 the FDA approved Vismodegib (tradename: ErivedgeTM, previously known as GDC-0449 and HhAntag691) a hedgehog signalling pathway inhibitor for the treatment metastatic basal cell carcinoma.
This is a novel first-in-class medicine, which for the first time modulates the hedgehog pathway. The target of Vismodegib is smoothened (SMO) (UniProt:Q99835, CanSAR:link). SMO is a class F (or class 6) G-Protein Coupled Receptor (GPCR) also known as the Frizzled/Smoothened class (pfam:PF01534). Smoothened is distinct and different to the previously 'drugged' GPCRs (which primarily target class A/class 1 receptors). The 3D-structures of smoothened or any of its homologues have not been characterised and are believed to differ from Class 1 GPCRs.
Vismodegib (Chembl: CHEMBL473417, ChemSpider:23337846 PubChem:CID24776445, IUPAC: 2-chloro-N-[4-chloro-3-(pyridin-2-yl)phenyl]-4-(methylsulfonyl)benzamide) is a synthetic small molecule drug, and is fully rule-of-five compliant. The molecular weight is 421.3, and has an XlogP of 3.8). Normal dosing is 150 mg per day (equivalent to 356 umol).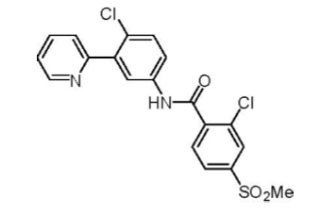
Vismodegib is a highly permeable compound with low aqueous solubility (BCS Class 2) - the single dose absolute bioavailability of vismodegib is 31.8%. Absorption of vismodegib is saturable - shown by the lack of dose proportional increase in exposure after a single dose of 270 mg or 540 mg vismodegib. The volume of distribution (Vd) of vismodegib ranges from 16.4 to 26.6 L, and plasma protein binding is greater than 99% - with binding observed to both human serum albumin (ALB) and alpha-1-acid glycoprotein (also known as Orosomucoid, ORM, AGP and AAG), it is also a substrate of the efflux transporter P-glycoprotein (P-gp). Greater than 98% of the total circulating drug-related components are the parent drug. Minor metabolic pathways of vismodegib in humans include oxidation, glucuronidation, and pyridine ring cleavage. Vismodegib and its metabolites are eliminated primarily by the hepatic route with 82% of the administered dose recovered in the feces and less than 5% in urine. The estimated elimination half-life (t1/2) of vismodegib is 4 days after continuous once-daily dosing, and 12 days after a single dose.
Vismodegib has a boxed warning for potential risk of causing embryonic death and serious birth defects.
The license for Vismodegib is held by Genentech.
Full Prescribing Information can be found here. -
The Future of Pharma in the UK

It has been difficult to avoid the news of the continuous downsizing of Pharma, and to those in the UK this has been especially painful recently. There is an excellent opinion piece here, written by Simon Campbell, ex-head of R&D at Pfizer, and also ex-President of the Royal Society of Chemistry. It was also interesting to see some of the comments from David Phillips (the current President of the of the Royal Society of Chemistry) that...
"the easy targets for new drugs... in the body... essentially have all been used up"
This is a major interest of ours, as regular readers will know; so expect a post on that exact subject soon.
-
New Drug Approvals 2012 - Pt. III - Axitinib (INLYTA®)

ATC Code: L01XE17Wikipedia: Axitinib
On Jan 27th 2012, the FDA approved Axitinib (also known as AG-13736, trade name: Inlyta), a kinase inhibitor, for the treatment of advanced renal cell carcinoma after failure of a first line systemic treatment.
Renal Cell Carcinoma (RCC) is a cancer of the lining of proximal convoluted tubules, the tiny tubes through which the blood is filtered, in the kidney. It is the most common type of kidney cancer in adults and is responsible for 80% of all kidney cancers (Cancer Research UK). Over 270,000 new cases of kidney cancers are diagnosed every year and the numbers are on the rise (CRUK).
Axitinib is a tyrosine kinase inhibitor, inhibiting all subtypes of the Vascular Endothelial Growth Factor Receptor (VEGFR), VEGRF1 (Uniprot:P17948; ChEMBL1868 ; canSAR), VEGFR2 (Uniprot:P35968; ChEMBL ; canSAR) and VEGFR3 (Uniprot:P35916 ; ChEMBL; canSAR).
VEGFRs are single-pass membrane receptors that have multiple extracellular Immunoglobulin-like domains involved in growth factor binding (the ligand is VEGF); and an intracellular Tyrosine Protein Kinase catalytic domain (pfam:PF07714). Axitinib inhibits this kinase domain (rough boundaries shown as sequence alignment)
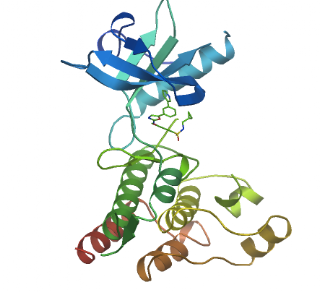
(PDB code: 1y6b; VEGFR2 kinase catalytic domain)
P17948 827 LKLGKSLGRGAFGKVVQASAFGIKKSPTCRTVAVKMLKEGATASEYKALMTELKILTHIGHHLNVVNLLGACTKQGGPLM 906 P35968 834 LKLGKPLGRGAFGQVIEADAFGIDKTATCRTVAVKMLKEGATHSEHRALMSELKILIHIGHHLNVVNLLGACTKPGGPLM 913 P35916 845 LHLGRVLGYGAFGKVVEASAFGIHKGSSCDTVAVKMLKEGATASEHRALMSELKILIHIGNHLNVVNLLGACTKPQGPLM 924 P17948 907 VIVEYCKYGNLSNYLKSKRDLFFLNKDAALHME-PKKEKMEPGLEQGKKP-RLDSVTSSESFASSGFQEDKSLSDVEEEE 984 P35968 914 VIVEFCKFGNLSTYLRSKRNEFVPYKTKGARFR-QGKDYVGAIPVDLKR--RLDSITSSQSSASSGFVEEKSLSDVEEEE 990 P35916 925 VIVEFCKYGNLSNFLRAKRDAFSPCAEKSPEQRGRFRAMVELARLDRRRPGSSDRVLFARFSKTEGGARRAS----PDQE 1000 P17948 985 DSDGFYKEPITMEDLISYSFQVARGMEFLSSRKCIHRDLAARNILLSENNVVKICDFGLARDIYKNPDYVRKGDTRLPLK 1064 P35968 991 APEDLYKDFLTLEHLICYSFQVAKGMEFLASRKCIHRDLAARNILLSEKNVVKICDFGLARDIYKDPDYVRKGDARLPLK 1070 P35916 1001 A-EDLWLSPLTMEDLVCYSFQVARGMEFLASRKCIHRDLAARNILLSESDVVKICDFGLARDIYKDPDYVRKGSARLPLK 1079 P17948 1065 WMAPESIFDKIYSTKSDVWSYGVLLWEIFSLGGSPYPGVQMDEDFCSRLREGMRMRAPEYSTPEIYQIMLDCWHRDPKER 1144 P35968 1071 WMAPETIFDRVYTIQSDVWSFGVLLWEIFSLGASPYPGVKIDEEFCRRLKEGTRMRAPDYTTPEMYQTMLDCWHGEPSQR 1150 P35916 1080 WMAPESIFDKVYTTQSDVWSFGVLLWEIFSLGASPYPGVQINEEFCQRLRDGTRMRAPELATPAIRRIMLNCWSGDPKAR 1159 P17948 1145 PRFAELVEKLGDLLQANVQQDGKDYI--PINAILTGNSGFTYSTPAFSEDFFK-ESISAPKFNSGSSDDVRYVNAFKFMS 1221 P35968 1151 PTFSELVEHLGNLLQANAQQDGKDYIVLPISETLSMEEDSGLSLPTSPVSCMEEEEVCDPKF--------HYDNTAGISQ 1222 P35916 1160 PAFSELVEILGDLLQGRGLQEEEEVCMAPRSSQ-SSEEGSFSQVSTMALHIAQADAEDSPPSLQRHSLAARYYNWVSFPG 1238 P17948 1222 L----------ERIKTFEELL---PNATSMFDDYQGDSSTLLASPMLKRFTWTDSKPKASLKIDLRVTSKS----KESGL 1284 P35968 1223 YLQNSKRKSRPVSVKTFEDIPLEEPEVKVIPDDNQTDSGMVLASEELKTL---EDRTKLSPSFGGMVPSKS----RESVA 1295 P35916 1239 CLARGAETRGSSRMKTFEEFPMTPTTYKGSVD-NQTDSGMVLASEEFEQI---ESRHRQESGFSCKGPGQNVAVTRAHPD 1314 P17948 1285 SDVSRPSF-CHSSCGHVSEGKRRFTYDHAELER----KIACCSPPPDY----NSVVLYSTPPI 1338 P35968 1296 SEGSNQTS--GYQSGYHSDDTDTTVYSSEEAELLKLIEIGVQTGSTAQILQPDSGTTLSSPPV 1356 P35916 1315 SQGRRRRPERGARGGQ-------VFYNSEYGELSEPSEEDHCSPSARVTFFTDNSY------- 1363There are many VEGF inhibitors in development, and several launched drugs also have activity against VEGFR (including Vandetanib, Sorafenib, Pazopanib and the broad spectrum inhibitor Sunitinib).Axitinib (Trade name: Inlyta®; IUPAC= N-methyl-2-[3-((E) 2-pyridin-2-yl-vinyl)-1H-indazol-6-ylsulfanyl]-benzamide; Canonical SMILES: CNC(=O)c1ccccc1Sc2ccc3c(\C=C\c4ccccn4)n[nH]c3c2 ; InChIKey=RITAVMQDGBJQJZ-FMIVXFBMSA-N); (ChEMBL1289926; canSAR)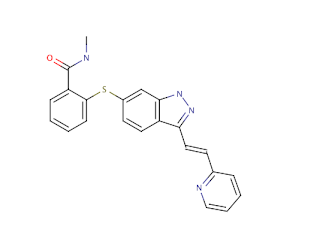
It has the molecular formula C22H18N4OS. Its molecular weight is 386.47, and has an AlogP of 4.49. Following single oral 5-mg dose administration, the median Tmax ranged between 2.5-4.1 hours.The mean oral bioavailability is 58%. Axitinib is highly bound (>99%) to human plasma proteins. The plasma half life (T1/2varies between 2.5 and 6.1 hours. It is metabolized primarily in the liver by CYP3A4/5 and to a lesser extent by CYP1A2, CYP2C19, and UGT1A1.
Full prescribing information can be found here.
Axitinib (Inlyta) is a product of Pfizer -
Estimates of Clinical Attrition
 I am the world's best procrastinator, always putting things off - today I've actually got a whole bunch of stuff done - the motivator for this was promising myself that once I'd done a big pile of stuff, I'd write something for the blog. Yes, I'm weird, but I find that writing this stream os ASCII characters on your screen right now, makes me think about the way to explain things to others, and also makes me ask the important question 'is this likely to be of any interest to anyone?'.
I am the world's best procrastinator, always putting things off - today I've actually got a whole bunch of stuff done - the motivator for this was promising myself that once I'd done a big pile of stuff, I'd write something for the blog. Yes, I'm weird, but I find that writing this stream os ASCII characters on your screen right now, makes me think about the way to explain things to others, and also makes me ask the important question 'is this likely to be of any interest to anyone?'.
Anyway, here's a toy analysis - Clinical attrition rates are the subject of a lot of analyses, the most rigorous ones usually involve the survey of a set of large pharma companies, and then analysis of the attrition. This is simple, and has led to various estimates of around 1:10 survival from phase 1 to launch to a 1:20 number. So 90 to 95% of compounds entering clinical trials fail, success probability is about 0.05. My hunch is that this is an underestimate - Logically, large pharma should be the best at pushing things through development, and if analysis is restricted to this best-in-class subset, surely the attrition of the remainder should be higher. Let's do a back of the envelope calculation, and let the data speak - or at least point us to the data we actually need to put together in order to do it right. I don't have time to point to some references for these numbers, that is for another day; remember, I'm only half way through my list of urgent things to do, or I will be in trouble.....
So, picking up on the discussion of kinase inhibitor attrition recently - about 20% of the USAN named compounds make it to market - this is roughly the same as the non-kinase set of drugs (data not shown). So this is 1:5 attrition (or 0.2 probability). Now, for the kinases, we have a pretty good set of data for all compounds that have entered clinical trials, and the ratio of non-USAN to USAN compounds is about 3:1, so using a steady state assumption (which is probably reasonable, data not shown, but the subject of a future post), gives a transition probability from entering clinical trials to non-proprietary name assignment of 0.25.
So overall, the attrition, from this small, but well characterised set is (0.2 * 0.25) which is 0.05, or 1 in 20. So things hang together quite well, and the number is sorta consistent with other analyses. Next some correlation with chemical properties.








