-
New Drug Approvals 2011 - Pt. XXVI - Icatibant (FirazyrTM)

ATCC: C01EB19Wikipedia: IcatibantOn the August 25th 2011, the FDA approved Icatibant (trade name: FirazyrTM), a bradykinin B2 receptor (B2R) antagonist indicated for the treatment of acute attacks of hereditary angioedema (HAE) in patients aged 18 or older.
HAE is a rare genetic disease and is caused by low levels of C1-esterase inhibitor (C1-INH), the major endogenous inhibitor and regulator of the protease plasma kallikrein and the key regulator of the Factor XII/kallikrein cascade. One component this cascade is the production of bradykinin by plasma kallikrein. During HAE attacks, disregulated activity of plasma kallikrein leads to excessive bradykinin production; bradykinin is a potent vasodilator, which s thought to be responsible for the characteristic HAE symptoms of localised swelling, inflammation and pain.
Icatibant treats the clinical symptoms of HAE attack by selective- and competitively binding, as an antagonist, to the B2 bradykinin receptor (B2R) (Uniprot: P30411; ChEMBL ID: CHEMBL3157; PFAM: PF00001), with similar affinity to bradykinin (1-10 nM for the B2R, while affinity for the B1R is 100-fold lower). Icatibant is the first in class agent against this target. The -tibant stem covers bradykinin antagonists.
B2R is a Rhodopsin-like receptor, 391 amino acid long, which belongs to the G protein-coupled receptor (GPCR) A3 family and is encoded by the BDKRB2 gene in humans. The amino acid sequence of B2R is:
>B2R MFSPWKISMFLSVREDSVPTTASFSADMLNVTLQGPTLNGTFAQSKCPQVEWLGWLNTIQ PPFLWVLFVLATLENIFVLSVFCLHKSSCTVAEIYLGNLAAADLILACGLPFWAITISNN FDWLFGETLCRVVNAIISMNLYSSICFLMLVSIDRYLALVKTMSMGRMRGVRWAKLYSLV IWGCTLLLSSPMLVFRTMKEYSDEGHNVTACVISYPSLIWEVFTNMLLNVVGFLLPLSVI TFCTMQIMQVLRNNEMQKFKEIQTERRATVLVLVVLLLFIICWLPFQISTFLDTLHRLGI LSSCQDERIIDVITQIASFMAYSNSCLNPLVYVIVGKRFRKKSWEVYQGVCQKGGCRSEP IQMENSMGTLRTSISVERQIHKLQDWAGSRQ
There are no known experimental structures of B2R, however there are several relevant homologous structures of other members of the rhodopsin-like GPCR family (see here for a current list).Icatibant is the third drug approved in the US to treat HAE attacks. Previous drugs include Ecallantide (approved in December 2009 under the trade name Kalbitor), which is a potent, selective, reversible inhibitor of plasma kallikrein, and C1-INH (approved in October 2009 under the trade name Berinert), which is a freeze-dried human C1-esterase inhibitor concentrate.
 Icatibant (IUPAC: (2S)-2-[[(3aS,7aS)-1-[2-[(2S)-2-[[(2S)-2-[[2-[[(4R)-1-[1-[2-[[(2R)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]-4-hydroxypyrrolidine-2-carbonyl]amino]acetyl]amino]-3-thiophen-2-ylpropanoyl]amino]-3-hydroxypropanoyl]-3,4-dihydro-1H-isoquinoline-3-carbonyl]-2,3,3a,4,5,6,7,7a-octahydroindole-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoic acid; ChEMBL ID: CHEMBL375218; PubChem: 71364; ChemSpider: 5293384) is a synthetic decapeptide that differs from bradykinin (a nonapeptide with an amino acid sequence RPPGFSPFR) at the amino acids' positions 3, 5, 7 and 8, which have been replaced by four non-natural amino acids, and also in one additional amino acid (a D-arginine) at the N-terminus of the bradykinin arginine at position 1. Thus, Icatibant amino acid sequence is D-Arg-Arg-Pro-Hyp-Gly-Thi-Ser-D-Tic-Oic-Arg. These modifications prevent Icatibant from being metabolised by major bradykinin-metabolizing enzymes, which makes it more stable that bradykinin. Icatibant has a molecular weight of 1304.5 Da.
Icatibant (IUPAC: (2S)-2-[[(3aS,7aS)-1-[2-[(2S)-2-[[(2S)-2-[[2-[[(4R)-1-[1-[2-[[(2R)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]-4-hydroxypyrrolidine-2-carbonyl]amino]acetyl]amino]-3-thiophen-2-ylpropanoyl]amino]-3-hydroxypropanoyl]-3,4-dihydro-1H-isoquinoline-3-carbonyl]-2,3,3a,4,5,6,7,7a-octahydroindole-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoic acid; ChEMBL ID: CHEMBL375218; PubChem: 71364; ChemSpider: 5293384) is a synthetic decapeptide that differs from bradykinin (a nonapeptide with an amino acid sequence RPPGFSPFR) at the amino acids' positions 3, 5, 7 and 8, which have been replaced by four non-natural amino acids, and also in one additional amino acid (a D-arginine) at the N-terminus of the bradykinin arginine at position 1. Thus, Icatibant amino acid sequence is D-Arg-Arg-Pro-Hyp-Gly-Thi-Ser-D-Tic-Oic-Arg. These modifications prevent Icatibant from being metabolised by major bradykinin-metabolizing enzymes, which makes it more stable that bradykinin. Icatibant has a molecular weight of 1304.5 Da.
Icatibant can be self-administrated through an injection in the abdominal area, thus providing a new option for the treatment of acute HAE attacks. The recommended dose is 30 mg administrated subcutaneously. In the case of inadequate response or recurrence of symptoms, additional dose may be administrated at intervals of at least 6 hours, with no more than 3 doses administrated in any 24-hour period (recommended daily dose equivalent 69.0 umol).
Following a 30 mg subcutaneous dose, the absolute bioavailability of Icatibant is ca. 97%, with a plasma clearance of 245 mL/min, a mean elimination half-life of 1.4 hours and a volume of distribution of 29 L. Icatibant is extensively metabolised by proteolytic enzymes to inactive metabolites that are primarily excreted in the urine, with <10% of the dose eliminated as unchanged drug. As would be anticipated for a peptide drug, Icatibant is not an inhibitor of major cytochrome P450 (CYP) isoenzymes and is not an inducer of CYP 1A2 and 3A4.
The full prescribing information can be found here.
Icatibant is marketed by Shire Human Genetic Therapies Inc. and the product website is www.firazyr.com (Since 2008, Icatibant has been approved for use in the European Union; the european SPC can be found here). -
New Drug Approvals 2011 - Pt. XXV crizotinib (Xalkori®)

ATCC: L01XE15Wikipedia: CrizotinibOn the August 26th 2011, the FDA approved crizotinib (trade name:Xalkori® Research code: PF-02341066), an anaplastic lymphoma kinase (ALK) inhibitor for the treatment of patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) that is ALK-positive as detected by an approved FDA-approved test.
Non-small cell lung carcinomas (NSCLC) are cancers of the epithelial cells in the lung and describe all types of lung carcinomas other than small cell carcinomas. NSCLCs make up 88% of all lung carcinomas (see Cancer Research UK pages) and comprise genetically distinct classes of cancer, the most common are: Lung adenocarcinoma, large cell carcinoma and squamous cell carcinoma. Across the NSCLC types, some tumors harbour an ALK fusion protein. The EML4-ALK fusion gene has been shown to be affect the outcome of drug response and cells show resistance to EGFR inhibitors.
Crizotinib is an orally-dosed receptor tyrosine kinase inhibitor with significant activity against ALK (UniProt:Q9UM73, CHEMBL4247), HGFR (also known as c-Met) (Uniprot:P08581 CHEMBL3717) and RON (UniProt:Q04912, CHEMBL2689), and it is the first in class agent against any of these targets. However, ALK is the main targeted protein in these ALK-dependent, EGFR inhibitor resistant cancers. ALK is a trans-membrane receptor tyrosine kinase with the regulatory domains being extracellular and the kinase catalytic domain intracellular. The amino-acid sequence of the full wild type kinase is:>ALK (full length wild type, kinase domain in red) MGAIGLLWLLPLLLSTAAVGSGMGTGQRAGSPAAGPPLQPREPLSYSRLQRKSLAVDFVV PSLFRVYARDLLLPPSSSELKAGRPEARGSLALDCAPLLRLLGPAPGVSWTAGSPAPAEA RTLSRVLKGGSVRKLRRAKQLVLELGEEAILEGCVGPPGEAAVGLLQFNLSELFSWWIRQ GEGRLRIRLMPEKKASEVGREGRLSAAIRASQPRLLFQIFGTGHSSLESPTNMPSPSPDY FTWNLTWIMKDSFPFLSHRSRYGLECSFDFPCELEYSPPLHDLRNQSWSWRRIPSEEASQ MDLLDGPGAERSKEMPRGSFLLLNTSADSKHTILSPWMRSSSEHCTLAVSVHRHLQPSGR YIAQLLPHNEAAREILLMPTPGKHGWTVLQGRIGRPDNPFRVALEYISSGNRSLSAVDFF ALKNCSEGTSPGSKMALQSSFTCWNGTVLQLGQACDFHQDCAQGEDESQMCRKLPVGFYC NFEDGFCGWTQGTLSPHTPQWQVRTLKDARFQDHQDHALLLSTTDVPASESATVTSATFP APIKSSPCELRMSWLIRGVLRGNVSLVLVENKTGKEQGRMVWHVAAYEGLSLWQWMVLPL LDVSDRFWLQMVAWWGQGSRAIVAFDNISISLDCYLTISGEDKILQNTAPKSRNLFERNP NKELKPGENSPRQTPIFDPTVHWLFTTCGASGPHGPTQAQCNNAYQNSNLSVEVGSEGPL KGIQIWKVPATDTYSISGYGAAGGKGGKNTMMRSHGVSVLGIFNLEKDDMLYILVGQQGE DACPSTNQLIQKVCIGENNVIEEEIRVNRSVHEWAGGGGGGGGATYVFKMKDGVPVPLII AAGGGGRAYGAKTDTFHPERLENNSSVLGLNGNSGAAGGGGGWNDNTSLLWAGKSLQEGA TGGHSCPQAMKKWGWETRGGFGGGGGGCSSGGGGGGYIGGNAASNNDPEMDGEDGVSFIS PLGILYTPALKVMEGHGEVNIKHYLNCSHCEVDECHMDPESHKVICFCDHGTVLAEDGVS CIVSPTPEPHLPLSLILSVVTSALVAALVLAFSGIMIVYRRKHQELQAMQMELQSPEYKL SKLRTSTIMTDYNPNYCFAGKTSSISDLKEVPRKNITLIRGLGHGAFGEVYEGQVSGMPN DPSPLQVAVKTLPEVCSEQDELDFLMEALIISKFNHQNIVRCIGVSLQSLPRFILLELMA GGDLKSFLRETRPRPSQPSSLAMLDLLHVARDIACGCQYLEENHFIHRDIAARNCLLTCP GPGRVAKIGDFGMARDIYRASYYRKGGCAMLPVKWMPPEAFMEGIFTSKTDTWSFGVLLW EIFSLGYMPYPSKSNQEVLEFVTSGGRMDPPKNCPGPVYRIMTQCWQHQPEDRPNFAIIL ERIEYCTQDPDVINTALPIEYGPLVEEEEKVPVRPKDPEGVPPLLVSQQAKREEERSPAA PPPLPTTSSGKAAKKPTAAEISVRVPRGPAVEGGHVNMAFSQSNPPSELHKVHGSRNKPT SLWNPTYGSWFTEKPTKKNNPIAKKEPHDRGNLGLEGSCTVPPNVATGRLPGASLLLEPS SLTANMKEVPLFRLRHFPCGNVNYGYQQQGLPLEAATAPGAGHYEDTILKSKNSMNQPGP
The ELM4-ALK translocation protduces a chimeric protein with the N-terminal part of ELM4 and the catalytic region of ALK. This chimeria is constitutively active causing unregulated proliferation (Soda et al)The structure of ALK in complex with crizotinib has been determined (2XP2)
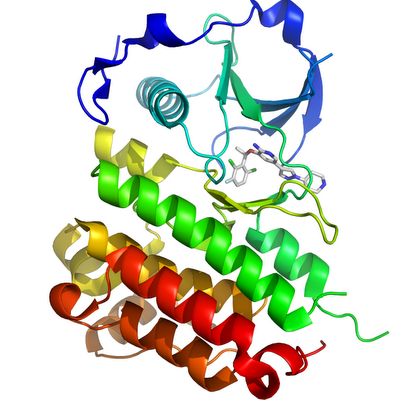
ALK and other proteins inhibited by crizotinib are members of the large protein kinase family, the target of several other recently approved drugs - with the approval of crizotinib there are now 12 US approved small molecule protein kinase inhibitors (
Imatinib, Gefitinib, Erlotinib, Sorafenib, Dasatinib, Sunitinib, Nilotinib, Lapatinib, Pazopanib, Vandetanib & Vemurafenib), with over an additional 300 protein kinase inhibitors that have progressed to clinical trials.
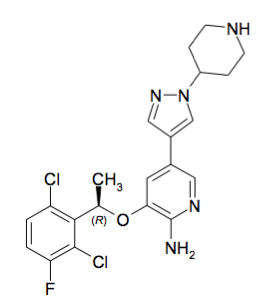
The molecular formula for crizotinib is C21H22Cl2FN5O, with a molecular weight of 450.34 Da. (IUPAC: (R)-3-[1-(2,6- Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine, Canonical SMILES:CC(C1=C(C=CC(=C1Cl)F)Cl)OC2=C(N=CC(=C2)C3=CN(N=C3)C4CCNCC4)N, InChI key:KTEIFNKAUNYNJU-GFCCVEGCSA-N, Chemspider:9801307, PubChem:11626560). It is fully rule of five compliant. Crizotinib has two ionizable centres with a pKa of 9.4 (for the piperidinium cation) and 5.6 (for the pyridinium cation). The experimental logD (octanol/water) at pH 7.4 is 1.65. Crizotinib is a chirally pure, synthetic small molecule drug.
Standard dosing of Crizotinib is 250mg twice daily (so a 500 mg daily dose - equivalent to 1,110 umol). The mean absolute bioavailability of Crizotinib is 43% following the administration of a single 250 mg oral dose. Crizotinib shows a median time to achieve peak concentration (Tmax) of 4 to 6 hours. Following crizotinib 250 mg twice daily dosing, steady state drug levels are reached within 15 days. The volume of distribution (Vss) of crizotinib is 1,772 L (following intravenous administration of a 50 mg dose) - so crizotinib is extensively distribution into tissues from the plasma; plasma protein binding (ppb) is 91%, and it is a substrate for P-glycoprotein (P-gp).
Crizotinib is predominantly metabolized by CYP3A4 and CYP3A5. The primary metabolic pathways in humans are oxidation of the piperidine ring to crizotinib lactam and O-dealkylation, followed by subsequent Phase 2 conjugation of O-dealkylated metabolites. Crizotinib is an inhibitor of CYP3A. The mean apparent plasma terminal half-life of crizotinib iss 42 hours, with an apparent clearance (CL/F) of 100 L/hr following a single dose, or 60 L/hr for the 250 mg twice daily standard dosing - this is most likely due to inhibition of CYP3A4/5.
Crizotinib is marketed by Pfizer, full prescribing information can be found here. -
ChEMBL-og NME Approval Monographs now on ChEMBL site
We've added a cumulative list of the ChEMBL-og drug monographs to the ChEMBL web interface. As new drugs, more specifically new molecular entities (NMES)) are approved (currently in the US) we write a short, consistent monograph on each approval. Coverage is from 2009 onwards. These typically appear a few days after the approval. We also provide a set of assignments (Rule of Five, Chiral, Black Box Warnings, etc.) for each drug. Some of these assignments are subjective, for example assigning a drug as natural product-derived is sometimes challenging.
We try to keep these current, so there will be inconsistency between the views through the ChEMBL interface and on this page, but this is currently inevitable in our versioned release of ChEMBL.
Any feedback on the view, data, etc. would be gratefully received. -
ChEMBL Release Scheduling
 We are currently reviewing our internal release process for ChEMBL, and have a question. When we do a release, there are sometimes changes to the schema (we generally detail these in the release notes of previous releases) and also changes to deprecated items (targets or compounds that have changed or disappeared). We now also have a series of sites that use the ChEMBL data to provide new services, and these will invariably break (often both in and outbound links) as loading and processing of the ChEMBL data happens in these other systems.
We are currently reviewing our internal release process for ChEMBL, and have a question. When we do a release, there are sometimes changes to the schema (we generally detail these in the release notes of previous releases) and also changes to deprecated items (targets or compounds that have changed or disappeared). We now also have a series of sites that use the ChEMBL data to provide new services, and these will invariably break (often both in and outbound links) as loading and processing of the ChEMBL data happens in these other systems.
One of the ideas we are considering is to have a 'pre-release' of the forthcoming release to interested parties during our normal one or two week long testing period. This could allow a smoother release schedule, and also help us with support issues after releases, but I'm sure have some further problems for us. So, if you run a resource, using ChEMBL and would like to discuss this with us, or if you have other views, we'd be happy to hear (mail link). -
PhD studentship Available for Oct 2012 Intake

We will be recruiting a PhD student to start in October 2012. Details of the application process can be found at www.embl.org/phdprogramme. The deadlines are important - you will need to register by December 1st 2011, with a final deadline of 12th December 2011.
There is a lot of exciting research in chemical biology at EMBL, and ideas for potential PhD projects within the chembl group include:
- Design of biopharmaceuticals using rule-based approaches.
- Drug design strategies to improve drug safety.
- Multi-scale indexing, visualisation and navigation of chemical structures.
- Drug attrition analysed using systems biology and simulation approaches.
However, we would be really keen to discuss your interests if you do apply. Contact us if you wish to discuss anything.
-
ChEMBL Identifiers

A few notes about the use and format of identifiers in ChEMBL:
Each of the major entity types within ChEMBL (documents, assays, compounds and targets) are assigned unique ChEMBL identifiers, which take the form of a ‘CHEMBL’ prefix followed immediately by an integer (e.g., CHEMBL25 is the compound aspirin, CHEMBL210 is the human beta-2 adrenergic receptor "target"). There is no distinction between the format of the identifier for different types of entities, but a given ChEMBL identifier will only ever be assigned to a single entity (i.e., CHEMBL25 will only ever be used for the compound aspirin and never for an assay, document or target). A lookup table is provided in the database, to resolve which identifiers correspond to which entity types.
ChEMBL identifiers are stable with respect to the entities they represent. For compounds (with known/defined structures), ChEMBL identifiers represent distinct compound structures, as defined by the standard InChI, e.g., CHEMBL25 represents: InChI=1S/C9H8O4/c1-6(10)13-8-5-3-2-4-7(8)9(11)12/h2-5H,1H3,(H,11,12). Therefore, two compounds reported in different papers but having the same standard InChI will be assigned the same ChEMBL ID.
These ChEMBL IDs will never be reassigned to a structure with a different standard InChI. However, since compounds may be reported or drawn incorrectly in the literature, it is sometimes necessary to alter the compound ChEMBL ID (structure) to which a particular bioactivity measurement links. In this case, the old (incorrect) ChEMBL identifier may be 'downgraded' in the database if no other data link to it. Downgraded compounds are not currently displayed on the live interface, but are retained in the database and the ChEMBL ID lookup table, and could be re-instated in future (with the same ChEMBL ID) if new data become available for them.
External identifiers for ChEMBL entities are also recorded in the database, where possible. For example, in addition to ChEMBL IDs and InChI/InChIKeys, all small molecule compounds with defined structures are assigned ChEBI identifiers. Where data are taken from other resources, the original identifiers are also retained (e.g., SIDs and AIDs for PubChem substances and assays, HET codes for PDBe ligands). PubMed identifiers or Digital Object Identifiers (DOIs) are stored for documents, and protein targets are represented by primary accessions from the UniProt database. -
New Drug Approvals 2011 - Pt. XXIV Vemurafenib (Zelboraf TM)

ATC code: L01XE15Wikipedia: Vemurafenib
On the August 17th 2011, the FDA approved Vemurafenib (trade name:Zelboraf TM Research code: PLX-4032, RG-7204 and RO-5185426), a BRAF kinase inhibitor for the treatment of patients with unresectable or metastatic melanoma carrying the mutant BRAFV600E.
Melanoma is a malignant tumor of melanocytes (skin cells that produce melanin) and is an aggressive disease responsible for an estimated 50,000 deaths worldwide. Over 50% of patients with advanced melanoma carry an activating mutation in the Serine/Theronine protein kinase: BRAF (V600E).
The MAPK signal transduction pathway is an important and frequently mutated pathway in cancer. A wide variety of growth factors signal through this pathway, via RAS and RAF proteins to cause cell proliferation. The activating mutation in BRAF causes activation of this pathway downstream of BRAF regardless of the presence of growth factor (the signalling pathway is 'dysregulated'). The protein of Vemurafenib is this mutant enzyme BRAFV600E (Uniprot:P15056(wt)), although it shows activity in vitro against other protein kinases including such as CRAF, ARAF, wild-type BRAF, SRMS, ACK1, MAP4K5 and FGR at similar affinities.
One side effect observed in nearly a quarter of patients is the paradoxical growth of cutaneous squamous cell carcinomas (cuSCC), a different, and less aggressive type of skin cancer. Intriguingly, this appears to be cause by activating the same pathway in normal cells of the same patient that carry a RAS mutation.

>BRAF MAALSGGGGGGAEPGQALFNGDMEPEAGAGAGAAASSAADPAIPEEVWNIKQMIKLTQEHIEALLDK FGGEHNPPSIYLEAYEEYTSKLDALQQREQQLLESLGNGTDFSVSSSASMDTVTSSSSSSLSVLPSS LSVFQNPTDVARSNPKSPQKPIVRVFLPNKQRTVVPARCGVTVRDSLKKALMMRGLIPECCAVYRIQ DGEKKPIGWDTDISWLTGEELHVEVLENVPLTTHNFVRKTFFTLAFCDFCRKLLFQGFRCQTCGYKF HQRCSTEVPLMCVNYDQLDLLFVSKFFEHHPIPQEEASLAETALTSGSSPSAPASDSIGPQILTSPS PSKSIPIPQPFRPADEDHRNQFGQRDRSSSAPNVHINTIEPVNIDDLIRDQGFRGDGGSTTGLSATP PASLPGSLTNVKALQKSPGPQRERKSSSSSEDRNRMKTLGRRDSSDDWEIPDGQITVGQRIGSGSFG TVYKGKWHGDVAVKMLNVTAPTPQQLQAFKNEVGVLRKTRHVNILLFMGYSTKPQLAIVTQWCEGSS LYHHLHIIETKFEMIKLIDIARQTAQGMDYLHAKSIIHRDLKSNNIFLHEDLTVKIGDFGLAT[V/E] KSRWSGSHQFEQLSGSILWMAPEVIRMQDKNPYSFQSDVYAFGIVLYELMTGQLPYSNINNRDQIIF MVGRGYLSPDLSKVRSNCPKAMKRLMAECLKKKRDERPLFPQILASIELLARSLPKIHRSASEPSLN RAGFQTEDFSLYACASPKTPIQAGGYGAFPVH
The structure of Vemurafenib complexed to BRAF is known (PDBe:3og7).
Vemurafenib
Vemurafenib (Molecular formula: C23H18ClF2N3O3S; IUPAC: N-(3-{[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]carbonyl}-2,4-difluorophenyl)propane-1-sulfonamide; Canonical smiles: CCCS(=O)(=O)Nc1ccc(F)c(C(=O)c2c[nH]c3ncc(cc23)c4ccc(Cl)cc4)c1F ; standard InChI: 1S/C23H18ClF2N3O3S/c1-2-9-33(31,32)29-19-8-7-18(25)20(21(19)26)22(30)17-12-28-23-16(17)10-14(11-27-23)13-3-5-15(24)6-4-13/h3-8,10-12,29H,2,9H2,1H3,(H,27,28) CHEMBL1229517, Chemspider:24747352, PubChem:CID 42611257). Vemurafenib is a synthetic small molecule drug, with no chiral centres, it has a molecular weight of 489.9 MWt and is fully rule of Five compliant.
Vemurafenib is orally administered as tablets, each tablet contains 240 mg of active compound - dosing is 960 mg twice daily (equivalent to 3920 umol). The bioavailability of vemurafenib has not been determined. Following oral administration of vemurafenib at 960 mg twice daily for 15 days to patients with metastatic melanoma, the median Tmax was approximately 3 hours. Vemurafenib is a moderate CYP1A2 inhibitor, a weak CYP2D6 inhibitor and is a CYP3A4 inducer, it is highly bound to serum albumin and alpha-1 acid glycoprotein (> 99% ppb). In treated patient populations the apparent volume of distribution is 106 L, the clearance is 31 L/day and the median half life is 57 hours. It is largely excreted in feces (94% of dose).
Vemurafenib is also notable in being arguably the first drug discovered and optimised using fragment soaking methods for initial lead discovery. Vemurafenib was discovered in the labs of Plexxikon.
Full US Prescribing information is here
Zelboraf is marketed by Hoffmann-La Roche Inc. -
New Drug Approvals 2011 - Pt. XXV Brentuximab vedotin (AdcetrisTM)

ATC code:L01XC12
Hodgkin's lymphoma is a cancer of cells derived from white blood cells called lymphocytes. In Hodgkin's lymphoma, the disease spreads from one lymph node group to another, and then leads to more systemic effects. Hodgkin's lymphoma can be treated with radiation therapy, chemotherapy or autologous hematopoietic stem cell transplantation. The occurrence of the disease shows two age peaks: the first in young adulthood (age 15–35) and the second in those over 55 years old
Brentuximab vedotin's molecular (for the antibody component of the ADC, Brentuximab) target is CD30 (Uniprot: P28908; canSAR ; PFAM: PF00020), which is expressed on activated T- and B-cells, and also is an established tumour marker. Endogenous CD30 ligands include TRAF1, TRAF2, TRAF3 and TRAF5. CD30 is a transmembrane protein, 577 amino acid long, containing three related copies of the TNFR_c6 domain (PFAM:PF00020). The structure of TRAF2 complexed to part of CD30 is known (see PDBe:1d01)

The antibody component Brentuximab binds to CD30 expressing cells, the the complex is then internalised, and the cytotoxic agent Monomethyl auristatin E (MMAE) which blocks cell division by preventing tubulin polymerisation. Auristatin is a marine natural product peptide derivative. There are approximately 4 copies of MMAE coupled to each antibody, and MMAE is linked through a protease activatable linker. The total molecular weight of Brentuximab vedotin is ca. 153 kDa. Brentuximab can therefore be considered to have two targets (CD30 and tubulin), and also to be a prodrug.
Brentuxumab vedotin is the second US approved antibody drug conjugate - the first being gemtuzumab ozagamicin (Mylotarg) which was approved in 2001, but subsequently withdrawn from the market. A significant number of ADCs are currently in clinical development.
Brentuximab is a human antibody. The antibody portion of Brentuximab vedotin has the sequence of two copies of:
>Brentuximab vedotin - heavy chain QIQLQQSGPEVVKPGASVKISCKASGYTFTDYYITWVKQKPGQGLEWIGWIYPGSGNTKY NEKFKGKATLTVDTSSSTAFMQLSSLTSEDTAVYFCANYGNYWFAYWGQGTQVTVSAAST KGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLY SLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSV FLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTY RVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTK NQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQG NVFSCSVMHEALHNHYTQKSLSLSPG >Brentuximab vedotin - light chain DIVLTQSPASLAVSLGQRATISCKASQSVDFDGDSYMNWYQQKPGQPPKVLIYAASNLES GIPARFSGSGSGTDFTLNIHPVEEEDAATYYCQQSNEDPWTFGGGTKLEIKRTVAAPSVF IFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLS STLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
The linker-MMAE has the following structure:

Brentuximab is administered intravenously, and the recommended dose is 1.8 mg/kg administered over 30 minutes every 3 weeks for a maximum of sixteen cycles doses. The terminal half-life (t1/2) is 4 to 6 days.
The full US prescribing information for Brentuximab vedotin can be found here. Adcetris™ is a product of Seattle Genetics









