Just a reminder of the first ChEMBL User Group, which will be held here on campus on Friday May 27th. Full details are on the LinkedIn group. If you are not a member, just send a join request! Attendees will each get a stick of ChEMBL rock.
The agenda, with speakers, titles, etc. will be posted shortly, both here and on the LinkedIn group.
So we can run some numbers, to get an idea of the scale of the issue, and then draw things together (in the next post) with a couple of things we are thinking of ourselves to do in ChEMBL.
Stereocenters
For a molecule stored in a database with a single undefined sp3 stereocenter, there are two possible distinct physical molecules (enantiomers), remember some properties are invariant w.r.t. the stereochemistry (e.g. logP) others aren't (e.g. binding energy to a receptor). As further undefined stereocenters are introduced, the number of possibilities increases as a simple combinatoric product. For three stereocenters, there are therefore 2**3 = 8 possibilities. There are a number of programs available to perform this stereo-enumeration - including stereoplex.
Tautomers
Enumeration of possible tautomers is a complex issue, and there has to be introduced the concept of an energy difference (which will reflect how frequent that tautomer occurs) - however the energy difference between two tautomers is crucially dependent on solvation and stabilisation of a particular tautomer by complexation in a binding site, so it is a not trivial task to treat this matter both comprehensively and accurately. There is probably an interesting average scaling of the number of 'reasonable tautomers' as a function of molecular weight for typical drug like molecules in a database such as ChEMBL (we just haven't looked yet). However, it is an area of active research, and there are many tools available to treat these systems - including MN.TAUTOMER.
Ionizable centers
As per tautomers there is the concept of reasonableness that needs to be applied here - in theory, benzene can act as both a base and an acid, but the pKb and pKa will be so outside the range encountered in what we currently understand as life-like conditions that it is irrelevant (the pKa of benzene is ~43). However, for molecules with 'regular' basic and acidic groups, there will be two (or more) states for each ionizable centers. There can be multiple states for some functional groups (e.g. polyprotic acids), such as phosphates which will have multiple pKa values, reflecting increasing levels of overall charge. These charge effects will greatly affect binding to a target. A further complicating factor is that the pKa of molecules is often perturbable by their molecular environment, and this shifting of 'standard' pKa values is often a defining feature of catalytic residues in enzymes.
However, for simple groups such as carboxylic acids, there are two states, and for simple aliphatic amines there are two states to be considered. These combine again in a combinatorial fashion, so a molecule with a simple basic and simple acidic center will have four possible states.
A key feature in dealing with the treatment of the ionization state of a molecule from a normalised chemical database is via prediction of the pKa/pKb of a molecule. There are many tools available to do this - including ACD/PhysChem Suite.
To further complicate things, the calculation of pKa/Pkb depends on the tautomers that the calculation is performed on.
Conformational flexibility
It is tempting to think of storing three-dimensional structures for molecules, since this information can be used in tasks such as docking of a library of rigid molecules to a receptor, or the generation of pharmacophores from a set of molecules that are known to bind to a receptor. However, in general the number of possible conformers is very large, and when combined with the additional complexities above of undefined stereocenters, tautomers, ionizable centers makes this a very challenging task. To give an idea of the complexity, two sp3-sp3 bonds will have three energy minima around that bond; as the number of rotatable bonds is increased, there will be an approximate combinatoric product, so for three independent sp3-sp3 rotatable bonds there will be 3**3 = 27 plausible conformers. Again these will have different energies (and therefore population frequencies), but again these energies are crucially dependent on tautomers, ionization and the solvent/receptor environment. It is complicated.
A very widely used estimate of the conformational flexibility of a molecule, and the implied entropic cost of ordering a flexible molecule on binding to a receptor, is the number of rotatable bonds.
Again there are many tools for the generation of reasonable three-dimensional conformers for a molecule - including CORINA.
Conclusion
So there is therefore a scale of complexity, molecules that are rigid, have no tautomeric forms, are not acids or bases and have completely defined (or no) stereochemistry are unambiguous, and can be safely used from a database like ChEMBL for things like docking. It is also possible to calculate a variety of descriptors for these molecules, and these calculations will be 'robust'. However it is important to appreciate that many property calculation methods require the selection of a single representative structure from the set of possibles.
Other molecules are more complex, and to obtain a physically relevant structure (or set of low energy structures), may require substantial processing/enumeration, the number of possible physical forms can easily extend into hundreds for drug-like molecules; for example, the simple molecule below, has 96 possible forms to consider for something like docking (and this ignores the very large number of conformational states from the 10 rotatable bonds in the structure, which alone are about 3**10 or over 59,000 'states').
An annotated form of the points of interest in this molecule is
In the next post, we will try and bring this together in the context of the ChEMBL database, and how we normalise our chemical structures on registration, and also how we calculate our descriptors, and some of the assumptions we make in this process.
The image above comes from the ever inspiring xkcd.
There is a publication just out covering some ideas around the prioritisation and development of drug discovery strategies for infectious diseases, involving collaborators from Dundee, UPenn and IBM Almaden. RAPID is one of those recursive acronyms, just like ZINC, or GNU.
Oh, and it's freely accessible, so grab a copy now!
Some time ago I posted, in an idle moment, the ChEMBL-og International H-value. The corresponding number now is 51 (of 143 distinct countries in total), so if you know any United Arab Emirate, Bangladesh, or Vietnam scientists (the countries who could realistically make a difference to this number) who would be interested in the sort of stuff we get up to, let them know!
Update. Looking at two analytics measures for the ChEMBL-og, both from Google. Google Analytics is consistently 30-35% down on the pageview count from the Blogger Analytics data - I guess the Blogger which is server side and doesn't require the js scripts of Google Analytics (and therefore is easy to suppress/block etc). I'd be interested in hearing other blog authors thoughts/experience.
I came across this interesting paper from earlier this year - "HIV proteinase inhibitors target the Ddi1-like protein of Leishmania parasites", published in FASEB J. HIV protease inhibitors were known to decrease levels of Leishmania in vivo, but the molecular target was not known. This paper shows that HIV-1 proteinase inhibitors are probably functional inhibitors of Ddi1 from Leishmania spp. Nelfinavir (the structure above) is a 440 nM IC50 inhibitor of L. major Ddi1 (and weaker against the human ortholog 3.3 uM). This is on the face of it, a lovely example of drug repositioning - the use of a drug for a new, and in this case, a non-obvious use.
HIV-1 PR (UniProt:Q9YQ34) and Ddi1 (UniProt:A4H334) are both aspartyl proteinases (Pfam:CL0129), share a common mechanism, and overall architecture (although HIV-PR is a homodimer, and Ddi1 is a single chain containing two 'copies' of the HIV-PR sequence). There is a human Ddi1 ortholog as well (UniProt:Q8WTU0), and poking around in UniProt shows there is a Tryp orthologue as well, but not an obvious Plasmo one - but I haven't really looked too hard, so far.
It is interesting to speculate what would be the most efficacious/potent launched/clinical stage HIV-1 PR inhibitor for the treatment of Leishmaniasis - and probably homology modelling and docking could allow a pretty good guess at this.
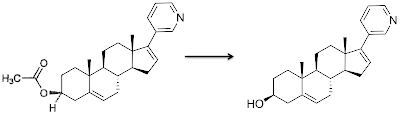
On 28th April 2011, the FDA approved Abiraterone acetate (Brand name ZytigaTM, NDA 202379) for the treatment of castrate-resistant prostate cancer (CRPC). Specifically, it is indicated for use in combination with prednisone for the treatment of patients with metastatic CRPC who have received prior chemotherapy containing docetaxel. Response biomarkers are amplified Androgen Receptor (AR); PTEN loss and hormone driven ERG rearrangement.
Abiraterone acetate (research code: CB-7630) is a steroid derivative with a chemical formula C26H33NO, (IUPAC: (3β) 17-(3-pyridinyl)androsta-5,16-dien-3-yl acetate; SMILES= (CC(=O)O[C@H]1CC[C@]2(C)[C@H]3CC[C@@]4(C)[C@@H](CC=C4c5cccnc5)[C@@H]3CC=C2C1 ); InChI=1S/C26H33NO2/c1-17(28)29-20-10-12-25(2)19(15-20)6-7-21-23-9-8-22(18-5-4-14-27-16-18)26(23,3)13-11-24(21)25/h4-6,8,14,16,20-21,23-24H,7,9-13,15H2,1-3H3/t20-,21-,23-,24-,25-,26+/m0/s1). It has molecular weight 391.55 and a LogP of 5.12. Abiraterone acetate is an ester prodrug which is converted in vivo to the active component, abiraterone.
Abiraterone acetate: (ChEMBLID:538435; canSAR link) Abiraterone: (ChEMBLID:522175; canSAR: link). Abiratrone has a terminal half-life (T1/2) of 12 (±5) hours.
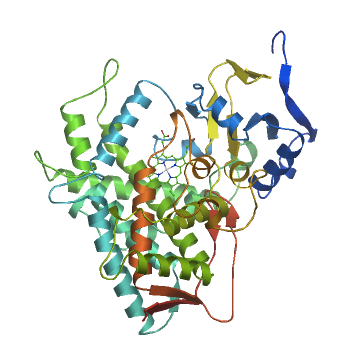
Abiraterone irreversibly inhibits 17-α-hydroxylase/C17,20-lyase (CYP17A1), an androgen biosynthesis enzyme expressed in testicular, adrenal, and prostatic tumor tissue. (Uniprot:P05093, canSAR link) and a member of the cytochrome P450 family of enzymes (PFAM:P450). Cyp17A1 normally catalyses a two step reaction: the conversion of pregnenolone and progesterone to their 17α-hydroxy- derivatives and then the formation of dehydroepiandrosterone (DHEA) and androstenedione, the precursors of testosterone. No structure of CYP17A1 itself exists, but it is homologous to other Cytochrome p450 enzymes such as CYP1A2 (PDBe:2hi4). Abiraterone's safety and effectiveness were established in a clinical study of 1,195 patients with late-stage castration-resistant prostate cancer who had received prior treatment with docetaxel chemotherapy, and showed improved survival. Zytiga is supplied in 250 mg tablets and the recommended dose is 1000 mg administered orally once daily in combination with prednisone 5 mg administered orally twice daily.
We have recently released a set of RESTful Web Services, which give users programmatic access to the ChEMBL data. During the Small Molecule Bioactivity course we hosted earlier in the year, and in a number of subsequent ChEMBL webinars, we've been asked if it is possible to use these new services in Pipeline Pilot, the short answer is yes.
To help users get started we have created a simple protocol, which you can download, use and modify (the license for this, for those interested is CC0). Remember to save the file on your disk - the link itself will look goofy in your browser. The protocol will retrieve data for a list of ChEMBLids and return a list of bioactivity and compound data in an html table.
We will be interested to hear how you get on, so tell us about any changes/enhancements you would make to the protocol, we're convinced it can be improved.
Over the coming months you can expect the set of ChEMBL Web Services to grow. We will keep you informed of any new additions via this blog, the website and the chembl-announce mailing list.
There are now a large number of GPCR structures known and freely available from resources such as PDBe. We are starting to add structural and binding site content to GPCR SARfari, and as part of this preparing a series of alignments, overlaps, etc.
Click this link for a structure-annotated sequence alignment (using joy) of current GPCR structures (as of April 2011). This representation is quite useful for identifying structurally variable regions, helix caps, and so forth. As you would expect for proteins of this divergence and degree of inherent flexibility, there are some ambiguities in the alignment.


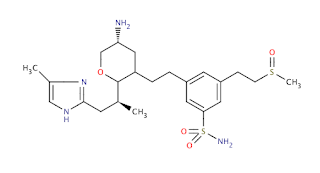 An annotated form of the points of interest in this molecule is
An annotated form of the points of interest in this molecule is




 Abiraterone's safety and effectiveness were established in a clinical study of 1,195 patients with late-stage castration-resistant prostate cancer who had received prior treatment with docetaxel chemotherapy, and showed improved survival. Zytiga is supplied in 250 mg tablets and the recommended dose is 1000 mg administered orally once daily in combination with prednisone 5 mg administered orally twice daily.
Abiraterone's safety and effectiveness were established in a clinical study of 1,195 patients with late-stage castration-resistant prostate cancer who had received prior treatment with docetaxel chemotherapy, and showed improved survival. Zytiga is supplied in 250 mg tablets and the recommended dose is 1000 mg administered orally once daily in combination with prednisone 5 mg administered orally twice daily. We have recently released a set of RESTful Web Services, which give users programmatic access to the ChEMBL data. During the Small Molecule Bioactivity course we hosted earlier in the year, and in a number of subsequent ChEMBL webinars, we've been asked if it is possible to use these new services in Pipeline Pilot, the short answer is yes.
We have recently released a set of RESTful Web Services, which give users programmatic access to the ChEMBL data. During the Small Molecule Bioactivity course we hosted earlier in the year, and in a number of subsequent ChEMBL webinars, we've been asked if it is possible to use these new services in Pipeline Pilot, the short answer is yes. 







