-
New Drug Approvals 2013 - Pt. XXIII Bazedoxifene (DUAVEE™)
wikipedia: bazedoxifeneATC code: G03XC02
On 3 October 2013, FDA approved a new drug, bazedoxifene in combination with estrogen (trade name DUAVEE™), for treatment of moderate-to-sever vasomotor symptoms (hot flashes) associated with menopause and the prevention of postmenopausal osteoporosis in women. Bazedoxifene reduces the risk of excessive growth of the uterus (endothermetrial hyperplasia) that can be caused by estrogen.
 Bazedoxifene (IUPAC name: 1-{4-[2-(Azepan-1-yl)ethoxy]benzy}-2-(4-hydroxyphenyl}-3-methyl-1H-indole-5-ol) is an indole based small molecule of molecular weight of 470.6 g/mol, polar surface area of 57.9, seven rotatable bonds, four hydrogen bond acceptors and one hydrogen bond donor. The compound is lipophilic with alogP 7.22, ApKa 10.12 and logD of 5.17. The compound is also known as WAY-140424, Bazedoxifene Acetate, Bazedoxifene, SID144206564, or SID124893775.Canonical Smiles: Cc1c(c2ccc(O)cc2)n(Cc3ccc(OCCN4CCCCCC4)cc3)c5ccc(O)cc15Standard InchI: InChI=1S/C30H34N2O3/c1-22-28-20-26(34)12-15-29(28)32(30(22)24-8-10-25(33)11-9-24)21-23-6-13-27(14-7-23)35-19-18-31-16-4-2-3-5-17-31/h6-15,20,33-34H,2-5,16-19,21H2,1H3
Bazedoxifene (IUPAC name: 1-{4-[2-(Azepan-1-yl)ethoxy]benzy}-2-(4-hydroxyphenyl}-3-methyl-1H-indole-5-ol) is an indole based small molecule of molecular weight of 470.6 g/mol, polar surface area of 57.9, seven rotatable bonds, four hydrogen bond acceptors and one hydrogen bond donor. The compound is lipophilic with alogP 7.22, ApKa 10.12 and logD of 5.17. The compound is also known as WAY-140424, Bazedoxifene Acetate, Bazedoxifene, SID144206564, or SID124893775.Canonical Smiles: Cc1c(c2ccc(O)cc2)n(Cc3ccc(OCCN4CCCCCC4)cc3)c5ccc(O)cc15Standard InchI: InChI=1S/C30H34N2O3/c1-22-28-20-26(34)12-15-29(28)32(30(22)24-8-10-25(33)11-9-24)21-23-6-13-27(14-7-23)35-19-18-31-16-4-2-3-5-17-31/h6-15,20,33-34H,2-5,16-19,21H2,1H3
Bazedoxifene is a selective estrogen receptor modulator (SERM) with a unique tissue and selectivity profile. Estrogen receptor (PDBe: 4iwf), is a transcription factor, which upon activation by a ligand, binds to DNA and regulates gene expression by mediating post-translational modification of histones and the associated transcriptional proteins.
 Bazedoxifene is also known to prevent bone loss and osteoporotic fractures in postmenopausal women. The drug, comes in the form of a tablet containing 0.45 mg estrogens and 20 mg bazedoxifene 20 mg. It is taken orally, once a day. Amongst other precautions, women taking DUAVEE should not take progestins, additional estrogens or additional estrogen agonist/antagonists. A detailed account of the prescribing information can be found here.
Bazedoxifene is also known to prevent bone loss and osteoporotic fractures in postmenopausal women. The drug, comes in the form of a tablet containing 0.45 mg estrogens and 20 mg bazedoxifene 20 mg. It is taken orally, once a day. Amongst other precautions, women taking DUAVEE should not take progestins, additional estrogens or additional estrogen agonist/antagonists. A detailed account of the prescribing information can be found here.
The license holders are Wyeth Pharmaceuticals, Inc. (a subsidiary of Pfizer Inc.,) based in Philadelphia, Pa. -
New Drug Approvals 2013 - Pt. XXII - Luliconazole (Luzu ™)
ATC Code: D01AC (incomplete)Wikipedia: Not AvailableChEMBL: CHEMBL2105689
On November 14th the FDA approved Luliconazole (trade name Luzu TM) for the treatment of skin fungal infections including ringworm. Luliconazole inhibits fungal synthesis of ergosterol, required for fungal cell membranes, by inhibiting the enzyme cytochrome P450 14-alpha-demethylase (P45014DM).
Target(s)
Like all azole antifungals, Luliconazole binds and inhibits fungal 14-alpha-demethylase (e.g. CHEMBL1681624, Uniprot Q96W81). P45014DM is a cytochrome P450 that catalyses the oxidative removal of the 14α-methyl group from eburicol to ergosterol. Azoles bind to the haem in P45014DM via the unprotonated N atom and occupy the active site as non-competitive inhibitors.
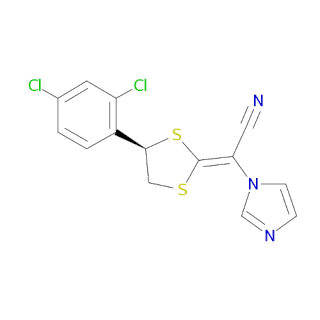 Luliconazole (CHEMBL2105689; Pubchem : 144206495 ) is a small molecule drug with a molecular weight of 354.3 Da, an AlogP of 4.03, 2 rotatable bonds, and no of 5 violations.
Luliconazole (CHEMBL2105689; Pubchem : 144206495 ) is a small molecule drug with a molecular weight of 354.3 Da, an AlogP of 4.03, 2 rotatable bonds, and no of 5 violations.
Canonical SMILES : Clc1ccc([C@@H]2CS\C(=C(\C#N)/n3ccnc3)\S2)c(Cl)c1InChi: InChI=1S/C14H9Cl2N3S2/c15-9-1-2-10(11(16)5-9)13-7-20-14(21-13)12(6-17)19-4-3-18-8-19/h1-5,8,13H,7H2/b14-12+/t13-/m0/s1
Each gram of Luzu Cream, 1% contains 10 mg of luliconazole in a white cream base.
Dosage
For treatment of Interdigital Tinea Pedis: Luzu Cream, 1% should be applied to the affected and immediate surrounding area(s) once a day for two weeks.
For treatment of Tinea Cruris and Tinea Corporis: Luzu Cream, 1% should be applied to the affected skin and immediate surrounding area(s) once a day for one week.
Drug InteractionsThe potential of luliconazole to inhibit cytochrome P-450 (CYP) enzymes 1A2, 2C9, 2C19, 2D6, and 3A4 was evaluated in vitro. Based on in vitro assessment, luliconazole at therapeutic doses, particularly when applied to patients with moderate to severe Tinea Cruris, may inhibit the activity of CYP2C19 and CYP3A4. However, no in vivo drug interaction trials have been conducted to evaluate the effect of luliconazole on other drugs that are substrates of CYP2C19 and CYP3A4.
Pregnancy
Luliconazole is classified as pregnancy category C. Animal reproduction studies have shown an adverse effect on the fetus and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite potential risks.
The license holder is Valeant Pharmaceuticals, the highlights of the prescribing information can be found here. -
New Drug Approvals 2013 - Pt. XXI - Eslicarbazepine Acetate (AptiomTM)

Wikipedia: Eslicarbazepine Acetate
On November 8th 2013, FDA approved Eslicarbazepine Acetate (tradename: Aptiom; research codes: Sep-0002093, BIA 2-093; ChEMBL: CHEMBL87992), a prodrug indicated as adjunctive treatment of partial-onset seizures associated with epilepsy.
Epilepsy is neurological disorder characterised by abnormal neuronal activity in the brain. Partial-onset seizures, as opposed to generalised seizures, affect initially only one part of the brain and, depending on the part of the brain that is affected, these seizures will present different symptoms.
Eslicarbazepine (ChEMBL: CHEMBL315985), the bioactive ingredient of the prodrug Eslicarbazepine Acetate, exerts its anticonvulsant activity by blocking the voltage-gated sodium channel (VGSC). VGSC has 3 distinctive states: the resting state, during which the VGSC is closed but responsive to a depolarisation impulse, the open state, during which the channel is open allowing the sodium ion to enter the cell, and the inactivated state, in which the channel is closed again but irresponsive to voltage changes. Eslicarbazepine binds and stabilises the inactive form of the VGSC, preventing its reversion to the resting form and limiting sustained repetitive neuronal firing.
VGSC (ChEMBL: CHEMBL2331043) is a single alpha-subunit with four repeat domains each containing six transmembrane segments. A 3D structure of the VGSC in an open conformation (PDBe: 4f4l) is shown below.

Eslicarbazepine Acetate is a synthetic small molecule with a molecular weight of 296.3 g.mol-1, an ALogP of 2.4, 3 hydrogen bond acceptors, 1 hydrogen bond donor, and therefore fully compliant with Lipinski's rule of five.
IUPAC: [(5S)-11-carbamoyl-5,6-dihydrobenzo[b][1]benzazepin-5-yl] acetate
Canonical Smiles: CC(=O)O[C@H]1Cc2ccccc2N(C(=O)N)c3ccccc13
InCHI: InChI=1S/C17H16N2O3/c1-11(20)22-16-10-12-6-2-4-8-14(12)19(17(18)21)15-9-5-3-7-13(15)16/h2-9,16H,10H2,1H3,(H2,18,21)/t16-/m0/s1
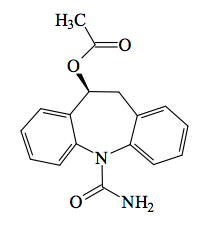
The recommended starting dosage of Eslicarbazepine Acetate is 400 mg once daily. After one week, the dosage should be increased to 800 mg once daily (recommended maintenance dosage). The maximum recommended maintenance dosage is 1200 mg once daily (after a minimum of one week at 800 mg once daily).
After oral administration, Eslicarbazepine Acetate is mostly undetectable, since it is extensively and rapidly metabolised by hydrolytic first-pass metabolism to its major active metabolite, Eslicarbazepine, corresponding to 91% of systemic exposure. Eslicarbazepine is highly bioavailable with an apparent volume of distribution of 61L for body weight of 70Kg, a relatively low plasma protein binding (< 40%) and an apparent half-life in plasma of 13-20 hours. Other minor active metabolites of Eslicarbazepine Acetate include (R)-Liscarbazepine and Oxcarbazepine, corresponding to 5% and 1% of systemic exposure, respectively. Eslicarbazepine Acetate metabolites are eliminated mainly by renal excretion, in the unchanged and glucuronide conjugated forms, with Eslicarbazepine and its glucuronide accounting for more than 90% of total metabolites excreted in urine.
The licensed holder of Eslicarbazepine Acetate is Sunovion Pharmaceuticals Inc. and the full prescribing information can be found here.
-
ADME SARfari: A tool for predicting and comparing cross-species ADME targets
 ADME studies are focused on understanding the disposition of a compound within an organism and the results of such studies play a critical role in the drug development process. ADME studies (more commonly referred to as pharmacokinetic or PK studies) are focused on 4 main areas: Absorption, Distribution, Metabolism and Excretion. More information on the PK measurement types can be found here.Comparisons of PK data across species is a potential problem drug researchers need to deal with, as model organism studies are the primary source of such data. For example, in an animal model study, which may be carried out on a compound as it passes through the drug development pipeline, is it meaningful to compare clearance or bioavailability data from a mouse or rat to human? Clearly there are many differences (physical, metabolic, genetic,..), which make answering these types of questions difficult. Building tools which guide researchers to potential answers or provide a better understanding of the inter-species differences are of great value - leading us nicely to the focus of this blog post.
ADME studies are focused on understanding the disposition of a compound within an organism and the results of such studies play a critical role in the drug development process. ADME studies (more commonly referred to as pharmacokinetic or PK studies) are focused on 4 main areas: Absorption, Distribution, Metabolism and Excretion. More information on the PK measurement types can be found here.Comparisons of PK data across species is a potential problem drug researchers need to deal with, as model organism studies are the primary source of such data. For example, in an animal model study, which may be carried out on a compound as it passes through the drug development pipeline, is it meaningful to compare clearance or bioavailability data from a mouse or rat to human? Clearly there are many differences (physical, metabolic, genetic,..), which make answering these types of questions difficult. Building tools which guide researchers to potential answers or provide a better understanding of the inter-species differences are of great value - leading us nicely to the focus of this blog post.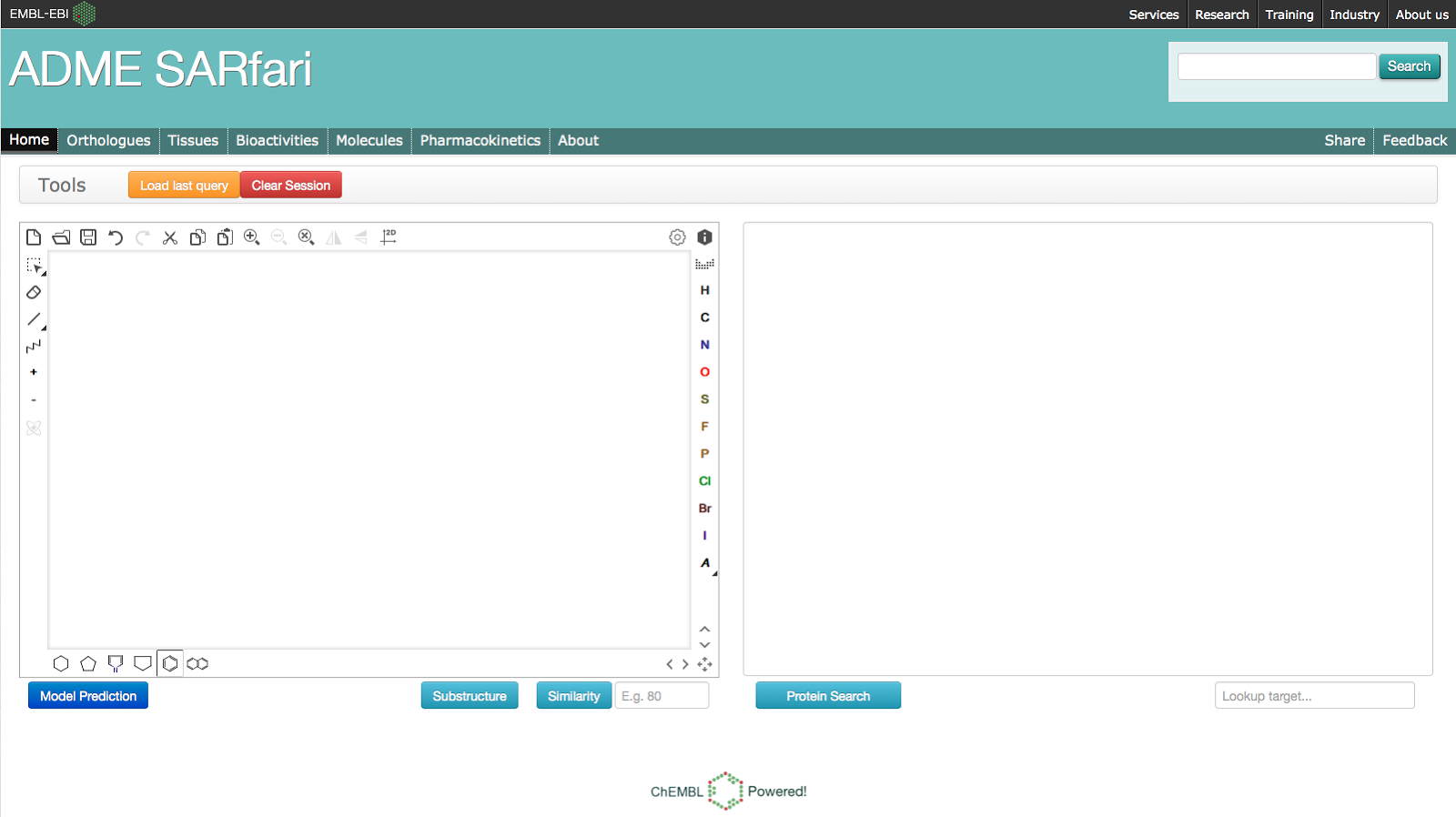
It turns out the ChEMBL database has a wealth of PK measurements data, which allows users to start asking ADME focused questions. You can access all of this data via the ChEMBL Interface or from one of our downloads, but in order to answer some of the more complex questions a significant amount of data processing first needs to take place. So to help the ChEMBL community get started analysing this data, we, in collaboration with our colleagues at GSK, set about building a new ADME focused system. The new system is called ADME SARfari and it aims to centralise all ADME data currently stored in ChEMBL and other related databases, as well as providing new tools to help interrogate the data.In order to build the system we have pulled data from a number of sources, which include:- ChEMBL - Bioactivity data, PK data and molecules.
- PharmaADME - An online resource providing a list ADME related human genes. We used this to build our primary list of ADME Targets, but have also added a couple of extra ones.
- The Human Protein Atlas - Protein expression data for human ADME Targets.
- ENSEMBL - Orthologue (using the Compara Service) and SNP data for ADME Targets.
- The Göttingen minipig and beagle dog genome predicted ADME Targets. GSK have sequenced the genomes of these two pharmaceutically significant species. More details on the genomes can be found here.
When you visit the site you will find it is divided into the following 7 sections:- Home - Allows user to initiate a compound or protein focused search.
- Orthologues - A table of ADME orthologues. The first column of the table corresponds to the human ADME targets and additional columns correspond to targets found in model organisms.
- Tissues - Protein expression data for human ADME targets.
- Bioactivities - Bioactivity data and PK measurements for all ADME related targets in the system, which are also found in the ChEMBL database.
- Molecules - Distinct set of ChEMBL molecules linked to ADME related targets via the bioactivity data.
- Pharmacokinetic - A cross species comparison of PK data for compounds found in the ChEMBL database (see red heatmap image at top).
- About - More details on how to use the system
By clicking on the links above, you can view and download all of the data associated within each of the sections. One exception is the Pharmacokinetics section, as there is too much data to display in the heatmap by default, so you must first initiate a search (we will come back to this later). Now that you have a better idea of what is in the system, what are the types of questions you can ask? Looking at the homepage you will see there are 2 ways to initiate a search of the system: Compound-initiated, using the chemical sketcher box and protein-initiated, using the BLAST or keyword search.Protein Search (Using BLAST)To run a BLAST search paste a sequence in the text box to the right on the homepage: BLAST search results will be displayed on the Orthologues page and only rows which contain hits to your search query will be returned:
BLAST search results will be displayed on the Orthologues page and only rows which contain hits to your search query will be returned: Clicking on the Tissues tab displays the protein expression levels of the human targets returned by the BLAST search:
Clicking on the Tissues tab displays the protein expression levels of the human targets returned by the BLAST search: Clicking on the Bioactivities tab returns all of the ChEMBL bioactivity data for targets returned by BLAST search:
Clicking on the Bioactivities tab returns all of the ChEMBL bioactivity data for targets returned by BLAST search: Clicking on the Molecules tab returns the distinct set of compounds currently displayed in the Bioactivities section:
Clicking on the Molecules tab returns the distinct set of compounds currently displayed in the Bioactivities section: Clicking on the Pharmacokinetics tab will provide a cross-species overview of the PK data in ChEMBL for the compounds currently displayed in the Molecules section (Note that not all compounds will have PK measurements):
Clicking on the Pharmacokinetics tab will provide a cross-species overview of the PK data in ChEMBL for the compounds currently displayed in the Molecules section (Note that not all compounds will have PK measurements): The following points help explain what the heatmap above is displaying:
The following points help explain what the heatmap above is displaying:- Each narrow row corresponds to a compound.
- Each column corresponds to a PK measurement in a specific organism.
- The PK measurements summarised in this view are Clearance (Cl), Cmax, Bioavailability (f), T1/2, Tmax and Volume of Distribution (Vd).
- The colour of each cell corresponds to a low, medium and high binned PK measurement, making it easier to compare values across species.
- The columns can be sorted by clicking on the header (see the second column, which corresponds to the Human Clearance data).
Compound SearchIt is also possible to search the system using a compound structure. Simply draw or paste a structure into the compound sketcher on the left of the homepage: You can then choose to run a substructure and similarity search. When you run the search you will first be taken Molecules section and you can then explore the other sections, which are all connected based on this initial set of compounds.Predictive Model SearchThe system also allows a user to predict which ADME protein target a molecule will interact with. The binding data (displayed in the Bioactivities section) has been used to build a multi-category naive Bayesian classifier. We will follow up with a more detailed post describing the model building process, but essentially the model is able to predict if a user-submitted molecule will interact with 133 of the ADME targets included in the system. The About page provides some additional data on the targets included in the model.To run a search against the predictive model, draw or paste a structure into the compound sketcher on the left of the homepage and hit the "Model Prediction" button. You will be taken to the Orthologues section, but only the rows which include a target predicted to interact with submitted molecule (coloured green) will be on display:
You can then choose to run a substructure and similarity search. When you run the search you will first be taken Molecules section and you can then explore the other sections, which are all connected based on this initial set of compounds.Predictive Model SearchThe system also allows a user to predict which ADME protein target a molecule will interact with. The binding data (displayed in the Bioactivities section) has been used to build a multi-category naive Bayesian classifier. We will follow up with a more detailed post describing the model building process, but essentially the model is able to predict if a user-submitted molecule will interact with 133 of the ADME targets included in the system. The About page provides some additional data on the targets included in the model.To run a search against the predictive model, draw or paste a structure into the compound sketcher on the left of the homepage and hit the "Model Prediction" button. You will be taken to the Orthologues section, but only the rows which include a target predicted to interact with submitted molecule (coloured green) will be on display: We hope you find the ADME SARfari system useful and if you have any questions please let us know.The ChEMBL Team
We hope you find the ADME SARfari system useful and if you have any questions please let us know.The ChEMBL Team -
New Drug Approvals 2013 - Pt. XX - Simeprevir (OlysioTM)

On November 22th 2013, the FDA approved simeprevir (Tradename: Olysio; Research Code(s): TMC-435; TMC435350), a Hepatitis C virus NS3/NS4A protease (HCV NS3/NS4A) inhibitor, for the treatment of chronic hepatitis C virus genotype 1 infection, in combination with peginterferon alfa and ribavirin.
Chronic hepatitis C is a prolonged infection that affects the liver and is caused by a small single-stranded RNA virus, which is transmitted by blood-to-blood contact. Chronic hepatitis C is normally asymptomatic, but may lead to liver fibrosis, and thus liver failure.
Simeprevir is an inhibitor of the hepatitis C virus (HCV) serine protease NS3/NS4A (ChEMBLID:CHEMBL2095231; Uniprot ID:A3EZI9, D2K2A8; Pfam:PF02907), a viral protein complex required for the proteolytic cleavage of the HCV encoded polyprotein (UniProt:P27958) into mature forms of the NS4B, NS5A and NS5B proteins. These proteins are involved in the formation of the virus replication complex, and therefore are vital to its proliferation. In a biochemical assay, simeprevir inhibited the proteolytic activity of recombinant genotype 1a and 1b HCV NS3/4A proteases, with median Ki values of 0.5 nM and 1.4 nM, respectively. However, in patients infected with the genotype 1a hepatitis C virus with an NS3 Q80K polymorphism, the effectiveness of simeprevir is slightly reduced, thus, screening for this polymorphism prior to the beginning of therapy is recommended, and alternative therapies should be considered.
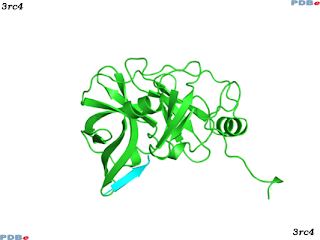
There are several protein structures known for HCV NS3 in complex with inhibitors, a typical entry is PDBe:3rc4, as expected from early genome annotation, the NS3 protease has a fold distantly related to the chymotrypsin-like family of serine proteases, and contains the classic Asp-His-Ser catalytic triad.
The -vir USAN/INN stem covers antiviral agents, and the substem -previr indicates it is a serine protease inhibitor. Simeprevir is the third approved agent to target HCV NS3/NS4A, following the approval of Merck's Boceprevir (q.v.) and Vertex's Telaprevir in 2011. Contrary to its predecessors, simeprevir is a natural derived compound, which requires a substantial lower dose (~16x less) for an effective response. It is also once-daily dosed, offering thus a promising alternative therapy for potential non-complying patients. Other compounds in this class in late stage clinical development/registration or earlier stages of development include Genentech's Danoprevir (RG-7227, ITMN-191), Bristol Myers Squibb's Asunaprevir (BMS-650032), Vaniprevir (MK-7009), Schering's Narlaprevir (SCH-900518), Achillion's Sovaprevir (ACH-0141625), Gilead's Vedroprevir (GS-9451), Ciluprevir (BILN-2061), ABT-450, BI-201335, IDX-320, MK-5172, BIT-225, VX-500, ACH-1625 and GS-9256.

Simeprevir (IUPAC Name: (2R,3aR,10Z,11aS,12aR,14aR)-N-(cyclopropylsulfonyl)-2-({7-methoxy-8-methyl-2-[4-(1-methylethyl)thiazol-2-yl]quinolin-4-yl}oxy)-5-methyl-4,14-dioxo-2,3,3a,4,5,6,7,8,9,11a,12,13,14,14a-tetradecahydrocyclopenta[c]cyclopropa[g][1,6]diazacyclotetradecine-12a(1H)-carboxamide; Canonical smiles: COc1ccc2c(O[C@@H]3C[C@@H]4[C@@H](C3)C(=O)N(C)CCCC\C=C/[C@@H]5C[C@]5(NC4=O)C(=O)NS(=O)(=O)C6CC6)cc(nc2c1C)c7nc(cs7)C(C)C; ChEMBL: CHEMBL501849; PubChem: 24873435; ChemSpider: 23331536; Standard InChI Key: JTZZSQYMACOLNN-VDWJNHBNSA-N) is a a natural product derived compound, with a molecular weight of 749.9 Da, 9 hydrogen bond acceptors, 2 hydrogen bond donors, and has an ALogP of 4.8. The compound is therefore not fully compliant with the rule of five.
Simeprevir is available as an oral capsule and the recommended daily dose is a single capsule of 150 mg. In HCV-infected subjects, the steady-state is reached after 7 days of once daily dosing and the mean steady-state AUC24 is 57469 ng.h/mL (standard deviation: 63571). Simeprevir should be administered with food, since food enhances its bioavailability by up to 69%. In vitro studies indicated that simeprevir is extensively bound to plasma proteins (greater than 99.9%).
The primary enzymatic system involved in the biotransformation of simeprevir in the liver is CYP3A. Therefore, co-administration of simeprevir with inhibitors or inducers of CYP3A may significantly alter the plasma concentration of simeprevir. In vitro studies indicated that simeprevir is a substrate of P-gp, and is transported into the liver by OATP1B1/3. Following a single oral administration of 200mg, the terminal elimination half-life of simeprevir is 10 to 13 hours in HCV-uninfected subjects and 41 hours in HCV-infected subjects. Elimination of simeprevir occurs via biliary excretion, and its metabolites are primarily excreted in feces.
As simeprevir is given as a component of a combination antiviral treatment regime with ribavirin and peginterferon alfa, there is a warning for embryofetal toxicity.
The license holder for OlysioTM is Janssen Pharmaceuticals, and the full prescribing information can be found here. -
New Year, New Job? Research Associate in Epidemiology at UCL (ChEMBL related)
As part of a collaboration the ChEMBL group are involved in with UCL, we are looking to appoint to a full time, three-year position in the Genetic Epidemiology Group, Institute of Cardiovascular Science, one of the component institutes of the UCL Faculty of Population Health Sciences.
The appointee will join an exciting programme of work funded by the UCL National Institute of Heath Research Biomedical Research Centre, through its High Impact Award scheme. The appointee will apply bioinformatic expertise to the late stage development of a new high density genotyping array designed to support drug target validation and related drug development issues; co-ordinate deployment of the array in a large consortium of highly-phenotyped cohort studies (the University College-London School of Hygiene-Edinburgh-Bristol consortium); undertake statistical analysis of the data; and play a leading role in writing manuscripts reporting findings arising from this work.
The post is based at UCL, but the work will involve collaboration with colleagues in Edinburgh, Bristol, the London School of Hygiene and Tropical Medicine, and the European Bioinformatics Institute at the Wellcome Trust Genome Campus in Hinxton, Cambridgeshire.
Key Requirements - The post would suit someone whose background is in genetic epidemiology, statistical genetics, or bioinformatics, particularly those whose expertise extends across two or more of these areas. We expect the appointee to have a PhD or equivalent degree. We are willing to consider applications from exceptional individuals who have only recently obtained their PhD degree, provided they can demonstrate the requisite skill. We place a strong emphasis on potential for independence and on supporting career development.
We particularly welcome female applicants and those from an ethnic minority, as they are under-represented within UCL at this level.More details and how to apply are here.
Closing Date - 12 Jan 2014jpo -
Conference: CEADD2014 - Modelling water in biological systems, London, March 2014
A one day conference entitled 'Modelling Water in Biological Systems' will be held at the School of Oriental and African Studies (SOAS) in London on Friday, 28 March, 2014. This meeting, organised by the MGMS, is the latest in the 'Cutting Edge Approaches to Drug Design (CEADD)' series.
In recent years, significant progress has been made in probing the role of water molecules in protein-ligand binding. Hydration is a crucial factor in understanding binding modes, ligand affinities and kinetics. Modelling tools are becoming available which may offer new insights in this exciting and evolving area of current research. This conference provides a timely overview of some of the main research avenues in this important field. -
ChEMBL Web Service Update 4: A Reminder

This post is to remind users of the ChEMBL Web Services that we will soon be changing the backend to use the new ChEMBL API. Since our initial announcement about the changes, which you can read about here, here and here, we have made some more changes and optimisations, which speed up the services significantly.
We thank everyone for feedback to date and urge anyone else who makes use of the ChEMBL Web Services to test the new version. Remember they are simple to test, just use the following temporary base URL and everything should work as if you are using the current live Web Services:
https://www.ebi.ac.uk/chemblws2
We would like to make the change in January, so please get in touch if you have any questions or experience any problems.
Once we have made the technology switch and happy that it is working in the wild as expected, we will be doing a complete review of the functionality offered by the current ChEMBL Web Services. So expected some big changes in 2014.






