-
Antibacterial Targets - Evidence for exclusion of targets for which host orthologues exist

One of the classic mantras for the genomics-based discovery of novel anti-bacterials is to ignore targets for which orthologues exist in the host (human) genome, but my hunch was that the majority of antibacterial mechanisms have clear host orthologues (they do, as you'll see below). I haven't come across any really simple papers supporting this dogma in the past, so decided to have a quick look this morning. I am the unwilling host for an oral bacterial infection myself at the moment, but I must stress that the throat above is not mine, but an anonymous one from Teh Interweb!
So, using a book I've just picked up at the ACS in Indy, I went through and did some quick analysis - the prose in the book is great, informal, and very very readable - buy it!
%T Antibacterial Agents: Chemistry, Mode of Action, Mechanisms of Resistance and Clinical Applications %A R.J. Anderson %A P.W. Groundwater %A A. Todd %A A.J. Worsley %I Wiley %D 2012 %O ISBN 978-0-470-97245-8
I went through, and at a drug class level, assigned the distinct mechanisms into three target classes.
- Orthologue of antibacterial target is present in humans.
- Orthologue of antibacterial target is absent in humans.
- Antibacterial acts through a non gene-derived target mechanism.
So, no real great evidence to focus on bacteria specific genes - in fact it's 2:1 in favour of targets for which orthologues exist in the host. The key would seem to be more exploiting physicochemical differences between bacterial and human cells (e.g. acidity), or exploit differences in the binding sites. I guess the dogma arose for a couple of reasons - firstly everyone knowns about penicillins, and secondly, it is an easy filter to apply bioinformatically, and finally it just seems like a perfectly sensible thing to do with respect to elimination of mechanism-based toxicity - and so is often done.
On this latter point, that of mechanism-based toxicity, this can be really important, but remember, a drug dosed to a human is not magically attracted only to relatives of the bacterial target, it will sample and equilibrate across all accessible binding sites of all proteins, and drugs will have side-effects and toxicity related to 1) binding to orthologues, 2) binding to paralogues, and 3) binding to anything else. A nice example of this is the paper from Science earlier this year on the side effects of sulphonamide antibacterials via inhibition of host sepiapterin reductase.
To be clear what I did here. Firstly - I used the chapters in the Antibacterial Agents book to define a class, so the graph can be plotted in many other ways - however, I was interested in distinct mechanisms. Secondly, several antibacterials don't target proteins, but various parts of the ribosome - these are gene derived, so count above as gene products, but they are not proteins (well they are riboproteins). Even if you strip these RNA targets out though (there are 5) the numbers are still not compelling for the need to avoid host target orthologues - the ratio would be 3:4 instead of 8:4.
What are the implications of removing this bacterial specific filter? Well that is more than a quick job on a Sunday morning and two cups of Lapsong Suchong - but my feeling is it might be quite significant.
jpo -
Seminar: Ruben Abgyan - The State of Docking, Modeling and Structure Based Molecular Discovery: GPCRs and Polypharmacology

We have Ruben Abagyan from Scripps visiting this coming Friday (27th of September 2013). Ruben will be well known to the computational chemistry and structural biology communities - he is giving a couple of talks, with the talk in the morning from 11am to noon "The State of Docking, Modeling and Structure Based Molecular Discovery: GPCRs and Polypharmacology" probably being of broad interest to many.
This will be an open seminar, but I will need to register any external attendees with security prior to Friday - so if you're in the Cambridge (UK) area, and wish to attend, please let me know by the end of Thursday. No web broadcasting or similar is possible for this. Sorry.
jpo
As an aside, Ruben is an EMBL Alumnus!
UPDATE - Please note change of time!! 11am to noon - Rosalind Franklin Room -
The Clinical Kinome in 2013

As part of a series of posts on the state of kinase inhibitors in formal clinical development, above is a mapping of the kinases for which there are potential drugs in trials. The figure was prepared by our collaborators Prson Gautam and Krister Winnerberg at FIMM, from the data in ChEMBL. -
New Drug Approvals 2013 - Pt. XIII - Dolutegravir (TivicayTM)
ATC code: J05AX12
On 12 August, the FDA approved a further drug for the treatment of HIV-1 infection, Dolutegravir (Tradename: Tivicay). Dolutegravir also known as S/GSK-1349575, is an HIV-1 integrase inhibitor. The drug has been approved for treatment of treatment-naïve as well as treatment-experienced HIV-infected adults including those who have been treated with other integrase inhibitors. In addition, Dolutegravir can be used for the treatment of children aged 12 years or older and weighing at least 40kg who have not been treated with integrase inhibitors, but are either treatment-naïve or treatment –experienced.
HIV, a lentivirus, infects vital cells in the human immune system such as helper T. cells (CD4+ T cells) and macrophages. The disease is responsible for millions of death every year, especially in Sub-Saharan Africa where treatment complications are enhanced by co-infection with tuberculosis and poverty. The approval of a new antiviral agent like Dolutegravir, will enhance treatment of the disease and improve the quality of people’s lives.
Dolutegravir is an inhibitor of HIV-1 integrase responsible for the insertion of the viral DNA into the host chromosomal DNA. The drug interferes with replication of HIV by preventing the viral DNA from assimilating into the genetic material of the human T cells. An example of a 3D structure of the enzyme’s core domain (PDBe: 3vqa) is shown below.

HIV-1 integrase (ChEMBLID: CHEMBL3471, UniProt Accession: Q72498) is an attractive target for drug design. It is one of three enzymes of HIV (others are Reverse Transcriptase and the Protease) that consists of three main domains with specific functions. The N-terminal domain characterized by the His2Cys2 motif chelates zinc, the core domain consists of the catalytic DDE motif important for the activity of the enzyme, and the C-terminal domain, with an SH3-like fold, that binds DNA nonspecifically. There are a variety of crystal structures of the different domains of HIV-1 integrase reported in PDBe (Protein Data Bank in Europe)
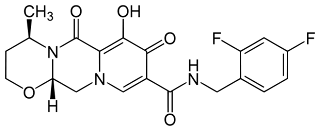
Dolutegravir , ChEMBLID: CHEMBL1229211 (C20H19F2N3O5, IUPAC Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,12,12a-tetrahydro-2H-pyrido[5,6]pyrazino[2,6-b][1,3]oxazine-9-carboxamide, Canonical smiles: CC1CCOC2N1C(=O)C3=C(C(=O)C(=CN3C2)C(=O)NCC4=C(C=C(C=C4)F)F)O) has two chiral centers, molecular weight of 419.12, 2 hydrogen bond donors, 6 hydrogen bond acceptors, 3 rotatable bonds, Polar surface area of 99.18 and alogP of 0.3. Dolutergravir is orally administered since it does not violate Lipinsik’s ‘Rule of Five’. The drug may be taken with or without food. For treatment-naïve or treatment-experienced with integrase transfer inhibitor (INSTI) – naïve adults and children the recommended dose is 50mg once. A dose of 50mg twice daily is recommended when dolutegravir is co-administered with potent UGT1A/CYP3A inducers like efavirenz, fosamprenavir/ritonavir, Tipranavir/ritonavir or rifampin.
The license holder for Dolutegravir is ViiV Healthcare, an HIV joint venture between GSK, Pfizer Inc and Shionogi. The full prescribing information can be found here.
-
Resources for Computational Drug Discovery - Wellcome Trust Course DEADLINE APPROACHING!
 It's that time of year again when the ChEMBL team and their collaborators come together to host the "Resources for Computational Drug Discovery" course.This course has been highly successful and well received over the past 3 years, and this year has no plans to be any different. It will be held here at the EBI campus in Hinxton, Cambridgeshire from the 9th - 13th December 2013.We will have speakers and instructors from institutes such as the University of California San Francisco, Institute of Cancer Research and University of Sheffield. The course will have both theoretical and practical sessions where the attendees will have a chance to apply what they have just learned.A provisional program can be found here.The deadline to sign up is looming (8th October) so click here to register and avoid disappointment!
It's that time of year again when the ChEMBL team and their collaborators come together to host the "Resources for Computational Drug Discovery" course.This course has been highly successful and well received over the past 3 years, and this year has no plans to be any different. It will be held here at the EBI campus in Hinxton, Cambridgeshire from the 9th - 13th December 2013.We will have speakers and instructors from institutes such as the University of California San Francisco, Institute of Cancer Research and University of Sheffield. The course will have both theoretical and practical sessions where the attendees will have a chance to apply what they have just learned.A provisional program can be found here.The deadline to sign up is looming (8th October) so click here to register and avoid disappointment!
Louisa -
New Drug Approvals 2013 - Pt. XIV - Tecfidera™

ATC Code: N07XX09 (2014)Wikipedia: Dimethyl FumerateChEMBL: CHEMBL2107333
On March 27th the FDA approved Dimethyl Fumarate (DMF, trade name TECFIDERA™) for the treatment of adults with relapsing forms of multiple sclerosis (MS). DMF and the metabolite, monomethyl fumerate (MMF), activate the Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway via inhibition of Kelch-like ECH-associated protein 1 (KEAP1, cytosolic inhibitor of Nrf2).
Target(s)The KEAP1 (CHEMBL2069156) is a naturally occuring cytosolic inhibitor of Nrf2 and DMF/MMF acts through chemical modification of KEAP1.
The NrF2 pathway is the primary cellular defence against the cytotoxic effects of oxidative stress. After translocation to the nucleus, Nrf2 heterodimerizes with MafF, MafG, and MafK. The combined heterodimer binds to antioxidant/electrophile response element (ARE/EpRE) and subsequently initiates transcription of these genes.
KEAP1 acts as the cytosolic anchor of Nrf2, sequestering Nrf2 in the cytoplasm during basal conditions. In addition KEAP1 contains a nuclear export signal and it is hypothesised to be the primary redox sensor. Thus DMF mediated inhibition of KEAP, leads to an increase of NrF2 translocation and increase in transcription of ARE/EpRE. This is hypothesized to be the working mechanism of DMF/MMF in MS. In addition, MMF has been shown to be an agonist of the nicotinic acid receptor (CHEMBL3785).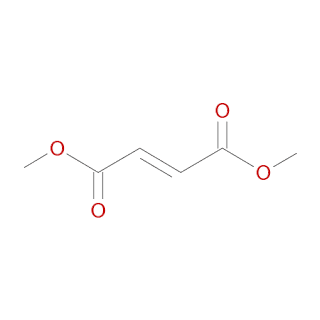 Dimethyl Fumarate (CHEMBL2107333 ; Chemspider : 553171; Pubchem : 99431554 ) is a small molecule drug with a molecular weight of 144.1 Da, an AlogP of 0.49 , 4 rotatable bonds and does not violate the rule of 5.
Dimethyl Fumarate (CHEMBL2107333 ; Chemspider : 553171; Pubchem : 99431554 ) is a small molecule drug with a molecular weight of 144.1 Da, an AlogP of 0.49 , 4 rotatable bonds and does not violate the rule of 5.
Canonical SMILES : COC(=O)\C=C\C(=O)OCInChi: InChI=1S/C6H8O4/c1-9-5(7)3-4-6(8)10-2/h3-4H,1-2H3/b4-3+
The recommended starting dose of TECFIDERA is 120 mg twice daily, for 7 days. Subsequently the dosage should be increased to a 240 mg twice daily maintenance dose. Tecfidera can be taken with or without food.
Dosage
Metabolism
In humans, dimethyl fumarate is extensively metabolized by esterases, which are ubiquitous in the gastrointestinal tract, blood, and tissues, before it reaches the systemic circulation. Further metabolism of MMF occurs through the tricarboxylic acid (TCA) cycle, with no involvement of the cytochrome P450 (CYP) system. MMF, fumaric and citric acid, and glucose are the major metabolites in plasma.
Excretion
Exhalation of CO2 is the primary route of elimination, accounting for approximately 60% of the TECFIDERA dose. Renal and fecal elimination are minor routes of elimination, accounting for 16% and 1% of the dose respectively. Trace amounts of unchanged MMF were present in urine.
The terminal half-life of MMF is approximately 1 hour and no circulating MMF is present at 24 hours in the majority of individuals. Accumulation of MMF does not occur with repeated dosing.
The license holder is Biogen Idec. the full prescribing information can be found here. -
ChEMBL_17 Released
We are pleased to announce the release of ChEMBL_17. This version of the database, prepared on 29th August 2013 contains:
- 1,519,640 compound records
- 1,324,941 compounds (of which 1,318,187 have mol files)
- 12,077,491 activities
- 734,201 assays
- 9,356 targets
- 51,277 documents
You can download the data from the ChEMBL FTP site. For more information please read the release notes.
Data changes since the last release:
Drug mechanism of action
For all FDA-approved drugs, information regarding the mechanism of action and associated efficacy targets has been curated from primary sources, such as literature and drug prescribing information. Targets have only been included for a drug if a) the drug is believed to interact directly with the target and b) there is evidence that this interaction contributes towards the efficacy of that drug in the indication(s) for which it is approved.
Metal-containing compounds
Structures for around 3200 metal-containing compounds have been removed from the database (though the bioactivity and other information for these compounds is retained). For more information, please see the previous blog posts: http://chembl.blogspot.co.uk/2013/08/removal-of-metal-containing-compounds.html
New data sets
Several new deposited/extracted data sets have also been included in the latest release: two deposited data sets from GlaxoSmithKine for Ghrelin receptor agonists and Motilin receptor agonists, a data set of the results of screening the MMV Malaria Box compound collection for activity against Schistosoma mansoni, two data sets screening the GSK PKIS compound collection for inhibition of luciferase activity, and finally pathology data from the Open TG-GATES project.
Interface changes since the last release:
Browse Drug Targets tab
A new tab has been created to show the new mechanism of action information for FDA approved drugs together with the references from which the information was obtained, and links to the relevant drug/target report card pages.
Document Report Card
A new table has been added to the document report card, showing other ChEMBL documents that are related to the current document. Pair-wise document similarity is assessed by two components. The first component is defined by whether a document cites or is referenced by the other. The second component is defined by the amount of overlap between the compounds and biological targets reported in the two respective documents. This overlap is quantified by the Tanimoto coefficient. Documents with the highest Tanimoto similarity scores to the query document are listed in this section. For example, the following page shows 5 additional ChEMBL documents that are deemed similar to the paper currently being viewed.
Database changes since the last release:
A number of new tables have been added to store the drug mechanism of action information (please see release notes and schema documentation for full details). In addition, a number of minor changes have been made to existing tables:
The PROTEIN_FAMILY_CLASSIFICATION table has been deprecated and replaced by a new hierarchical version: PROTEIN_CLASSIFICATION.
The MOLREGNO field has been removed from the ATC_CLASSIFICATION table and moved to a new mapping table: MOLECULE_ATC_CLASSIFICATION.
The MOLFORMULA field has been moved from the COMPOUND_STRUCTURES table to the COMPOUND_PROPERTIES table (and renamed).
The ChEMBL Team
-
New Drug Approvals 2013 - Pt. XII - Technetium Tc 99m Tilmanocept (LymphoSeekTM)
ATC code: V09IA09
On March 13th 2013, the FDA approved Technetium Tc 99m Tilmanocept (LymphoSeekTM), a radioactive diagnostic agent indicated for lymphatic mapping with a hand-held gamma counter to assist in the localisation of lymph nodes draining a primary tumour site in patients with breast cancer or melanoma.
Melanoma is a malignant skin tumour, which, although rather uncommon, causes 75% of skin cancer related deaths. Breast cancer accounts for almost 23% of all cancers in women and in 2008 caused 13.7% of cancer related deaths in women. Lymph nodes drain lymphatic fluid coming from tissues, if the tissues contain a tumour, the node will retain cancer cells coming from it. By removing and analysing the lymph node, precious informations can be obtained regarding the spread of the tumour.
Technetium Tc 99m Tilmanocept (ChEMBL: CHEMBL2108726) acts by accumulating in lymphatic tissue and selectively binding to mannose binding receptor (CD206, ChEMBL: CHEMBL2176854, Uniprot:P22897) found on macrophage and dendritic cells membrane. In vitro studies show that Technetium Tc 99m Tilmanocept binds to the human mannose binding receptor with an affinity of Kd = 2.76 x 10-11 M. Clinical studies show that it accumulates in lymph nodes within 10 min and up to 30 hours after the injection.
The PDBe entry (PDBe: 1egg) for a crystal structure of the macrophage mannose receptor is shown below.

Tilmanocept is a macromolecule composed of multiple units of diethylenetriaminepentaacetic acid (DTPA) and mannose, each covalently bonded to a 10 kDa backbone of dextran. The DTPA acts as a chelating agent for labelling with Technetium 99m (Tc 99m), while mannose, a naturally occurring sugar, acts as a target ligand. The active component is radioactive Tc 99m, a synthetic element widely used in nuclear medicine that decays with a half-life of 6 hours emitting Gamma-2 photons. The molecular formula of Technetium Tc 99m Tilmanocept is [C6H10O5]n.(C19H28N4O9S99mTc)b.(C13H24N2O5S2)c.(C5H11NS)a. It contains 3-8 conjugated DTPA molecules (b); 12-20 conjugated mannose molecules (c) with 0-17 amine side chains (a) remaining free. The calculated average molecular weight of Tilmanocept ranges from 15,281 to 23,454 g/mol.

The recommended dose of Technetium Tc 99m Tilmanocept is 18.5 MBq (0.5 mCi) as a radioactivity dose and 50 mcg as a mass dose, administered via injection at least 15 minutes prior the lymph node mapping. Technetium Tc 99m Tilmanocept has a half-life at the injection site of 1.75 to 3.05 hours.
LymphoSeek is produced by Navidea Biopharmaceuticals, Inc. Full prescribing information can be found here.













