On March 20th 2013, FDA approved Gadoteric Acid (as the meglumine salt; tradename: Dotarem; research code: P 449; CHEMBL: CHEMBL2219415), a gadolinium-based contrast agent (GBCA) indicated for intravenous use with magnetic resonance imaging (MRI) in brain (intracranial), spine and associated tissues of patients ages 2 years and older, to detect and visualize areas with disruption of the blood brain barrier (BBB) and/or abnormal vascularity of the central nervous system (CNS).
When placed in a magnetic field, Gadoteric Acid develops a magnetic moment. This magnetic moment enhances the relaxation rates of water protons in its vicinity, leading to an increase in signal intensity (brightness) of tissues. Gadoteric Acid enhances the contrast in MRI images, by shortening the spin-lattice (T1) and the spin-spin (T2) relaxation times.
Other GBCAs have already been approved by FDA for use in patients undergoing CNS MRI and these include Gadopentetate Dimeglumine (approved in 1988 under the tradename Magnevist; ChEMBL: CHEMBL1200431; PubChem: CID55466; ChemSpider: 396793), Gadoteridol (approved in 1992 under the tradename Prohance; ChEMBL: CHEMBL1200593; PubChem: CID60714; ChemSpider: 54719), Gadodiamide (approved in 1993 under the tradename Omniscan; ChEMBL: CHEMBL1200346; PubChem: CID153921; ChemSpider: 135661), Gadoversetamide (approved in 1999 under the tradename Optimark; ChEMBL: CHEMBL1200457; PubChem: CID444013; ChemSpider: 392041), Gadobenate Dimeglumine (approved in 2004 under the tradename Multihance; ChEMBL: CHEMBL1200571; PubChem: CID49799998; ChemSpider: 25046318) and Gadobutrol (approved in 2011 under the tradename Gadavist; ChEMBL: CHEMBL2218860; PubChem: CID15814656; ChemSpider: 26330337).
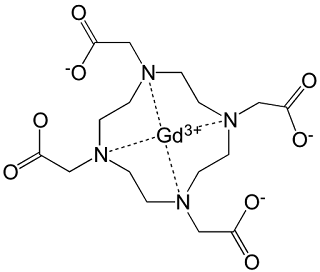
Gadoteric Acid is a macrocyclic ionic contrast agent, consisting of the chelating agent DOTA and gadolinium (Gd3+).
IUPAC: gadolinium(3+);2-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetic acid
Canonical Smiles: [Gd+3].OC(=O)CN1CCN(CC(=O)[O-])CCN(CC(=O)[O-])CCN(CC(=O)[O-])CC1
InChI: InChI=1S/C16H28N4O8.Gd/c21-13(22)9-17-1-2-18(10-14(23)24)5-6-20(12-16(27)28)8-7-19(4-3-17)11-15(25)26;/h1-12H2,(H,21,22)(H,23,24)(H,25,26)(H,27,28);/q;+3/p-3
The recommended dose of Gadoteric Acid is 0.2 mL/kg (0.1 mmol/kg) body weight administrated as an intravenous bolus injection at a flow rate of approximately 2 mL/second for adults and 1-2 mL/second for pediatric patients. Gadoteric Acid has a volume of distribution of 179 mL/kg and 211 mL/kg in female and male subjetcs, respectively, roughly equivalent to that of extracellular water, and an elimination half-life of about 1.4 hr and 2.0 hr in female and male subjects, respectively. Gadoteric Acid does not undergo plasma protein binding and it is not known to be metabolized. It is excreted primarily in the urine with 72.9% and 85.4% eliminated within 48 hours in female and male subjects, respectively. In healthy subjects, the renal and total clearance rates are comparable, with a renal clearance of 1.27 mL/min/kg and 1.40 mL/min/kg in female and male subjects, respectively, and a total clearance of 1.74 mL/min/kg and 1.64 mL/min/kg in female and male subjects, respectively.
All GBCAs, including Gadoterate Meglumine, carry a boxed warning about the risk of nephrogenic systemic fibrosis (NSF), a condition associated with the use of GBCAs in certain patients with kidney disease.
The license holder for Gadoterate Meglumine is Guerbet LLC and the prescribing information can be found here (Gadoteric Acid is also approved in Europe and the SPC can be found here).
There are more and more software libraries being ported to JavaScript. The best example is JavaScript/HTML5 Citadel demo of the Unreal Engine. So why not to try with some chemical stuff? One of the most important chemical software libraries is IUPAC InChi. It's also extremely hard to reimplement as it's written in low-level, functional-style C. On the other hand it's just a few headers and source files, without any dependencies so it's a perfect use case for Emscripten.
Emscripten 'is an LLVM-to-JavaScript compiler'. It can be used as a drop-in replacement for standard tools such as gcc or make. Recently it got support for asm.js - optimizable, low-level subset of JavaScript.
I wasn't the first to come up with this idea - one of our local heroes, Noel O'Boyle wrote a set of articles about translating the InChI code into JavaScript on his blog. I didn't know about his work during my experiments, which is good, because I took slightly different approach and came up with different results:
I decided to compile inchi-1 binary (by exposing its main function) not the library, because, according to readme file in InChI distribution package, the binary 'does extensively check the input data and does provide diagnostic concerning input structure' so it's the only tool that can be used as an InChi generator with 100% guarantee of having correct results for all input files.
The resulting JavaScript code (emscripten generated html with embedded JS) weighted 2.8 MB and 732 kB after zip compression (all modern servers and browsers support compressed files). The original inchi-1 binary is about 1.1 MB large so this sounds reasonable.
Of course there are some drawbacks of my approach - the most obvious one is IO. inchi-1 is command line tool expecting a file or plain text as input and printing some text to stdoutand stderr. JavaScript doesn't have any standard input or output. This means that this behavior must be somehow mapped to browser environment. Emscripten maps output to specific textarea element which is reasonable. On the other hand any request for user input is mapped to javascript prompt window. This prompt can accept one line of text at time. Molfiles contain many lines so putting a molfile line by line is tedious.
The solution to this problem would be adding a file input to the webpage and accessing it via Javascript Blob interface. Having the files selected allocating some memory in Emscripten using hints from this SO question and pass it to process_single_input function from inchimain.c file (this should be exported instead of main).
So far I haven't solved the last issue. You can check proof-of-concept here. To use it, open link in your browser, open javascript console (Control-Shift-K on Firefox, Control-Shift-J on Chrome), then type (as two separate commands, pressing Enter after each one):
bla = Module.cwrap('process_single_input', 'string', 'string')
bla('bla -STDIO')
After that, the standard javascript prompt will pop up. You have to copy there your mol file - line by line. If the line should be empty (usually 1st and 3rd lines are) just press enter in the input box. After last line (M END) hit cancel instead of OK. Then select a checkbox suppressing all further popups and press OK. If you entered all mol file lines correctly you will see the result!
Don’t we just love the fact that these days so much
bioactivity data is freely available at no cost (to the end user)? I think we do. The
more, the better. So, what would your answer be if someone asked you if you
consider it to be a good idea if they would deposit some of their unpublished bioactivity
data in ChEMBL? My guess is that you would be all in favour of this idea. 'Go
for it', you might even say. On the other hand, if the same person would ask you
what you think of the idea to deposit some of ‘your bioactivity data’ in ChEMBL
the situation might be completely different.
First and foremost you might respond that
there is no such bioactivity data that you could share. Well let’s see about
that later. What other barriers are there? If we cut to the chase then there is
one consideration that (at least in my experience) comes up regularly and this
is the question: 'What’s in it for me?' Did
you ask yourself the same question? If you did and you were thinking about
‘instant gratification’ I haven’t got a lot to offer. Sorry, to disappoint you.
However, since when is science about ‘instant gratification’? If we would all
start to share the bioactivity data that we can share (and yes, there is data
that we can share but don’t) instead of keeping it locked up in our databases
or spreadsheets this would make a huge difference to all of us. So far the main
and almost exclusive way of sharing bioactivity data is through publications
but this is (at least in my view) far too limited. In order to start to change
this (at least a little bit) the concept of ChEMBL supplementary bioactivity
data has been introduced (as part of the efforts of the Open PHACTS project, http://www.openphacts.org).
Here is how it works: If you have unpublished bioactivity
data that has been generated in an assay that can be found in ChEMBL (since the
publication where the assay is described is also in ChEMBL), you can now
deposit this data in ChEMBL (see http://dx.doi.org/10.6019/CHEMBL2094195 for an example). The obvious
situation would be one where only a subset of the results have been reported in
the publication but there are many more results (e.g. inactives). If you work
in an industrial setting and might feel that you are not be in a position to
release additional chemical structures you could think about depositing
bioactivity data for compounds in (older) patents. Or you have reported
bioactivity data in a poster. These are only examples and there are many more
opportunities. In some cases we might explore new territory and the progress
might be slow, but if we don’t try new things we are stuck with what we have. 'Do
we really want this?' I hope the answer is no. So, let’s not focus on ‘instant
gratification’ but help to grow the body of freely available bioactivity data
by contributing to ChEMBL supplementary bioactivity data. If we could just give
it a go it might make a difference. The concept might be quite restricted (e.g.
the assay needs to be published) but we need to start somewhere. If you want to
find out more about ChEMBL supplementary bioactivity data why not drop ChEMBL
Help a line (chembl-help@ebi.ac.uk) and put ‘ChEMBL supplementary bioactivity
data’ in the subject field. And don’t worry, you are not committing yourself by
wanting to know more.
ChEMBL, and the whole world of drug discoverers, is looking forward to hearing from you.
The map above shows the google analytics map view of the users of the ChEMBL-og. Being the completist that I am, it always bugs me, that there are still quite a few places that don't know about the ChEMBL-og. So if you have some friends in those lonely grey countries, such as Greenland, North Korea, Mali, Niger, Chad, Gabon, Central African Republic, DR Congo, etc. Tell them about the ChEMBL-og!
Our friendly neighbours at The Babraham Institute are looking for a PhD candidate to work on systems pharmacology models, as
part of a collaboration between the Le Novère (Babraham), the Hermjakob (EMBL-EBI) and the pharmaceutical company GlaxoSmithKline. The Le Novère groupuses quantitative computational models to understand cellular and
molecular processes, and develop community services that facilitate research in computational systems
biology (http://lenoverelab.org).
One of the major challenges of drug discovery is to demonstrate the efficacy of a potential new
drug. This goes beyond the development of a potent molecule - it also implies a good
understanding of the biological context, how it relates to a particular disease, and the drug's
mechanism of action. The availability of relevant Systems Pharmacology models can therefore have
a significant impact. The most comprehensive repository of Systems Biology models in machine
readable language is BioModels Database, created by Le Novère and maintained at the EBI. In spite
of its extensive collection, BioModels Database only covers a fraction of the Systems Pharmacology
models described in the literature. In addition, no analysis has been performed on how they map
to druggable targets and/or disease mechanisms.
The candidate will:
Use state-of-the-art
text-mining methods to extract and analyse the space of Systems Pharmacology models currently
described in the literature, with particular emphasis to their relevance to druggable targets and
disease mechanisms;
Identify the models offering the best opportunities for the discovery of
new drugs, and incorporate them into BioModels Database;
Explore and assess the
applicability of those models to real drug development cases, evaluating their quality, advantages,
caveats, overlaps, gaps and impact on the demonstration of drug efficacy against specific
indications.
The candidate must have an extensive knowledge of molecular biology and pharmacology, and
solid basis in numerical analysis and statistics. Advanced familiarity with data representation and
programming skills will also be desirable.
Thiele I et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013 Mar
3. Online advance publication.
Cucurull-Sanchez L et al. Relevance of systems pharmacology in drug discovery. Drug Discov Today.
2012 17: 665-670
Le Novère N et al. BioModels Database: a free, centralized database of curated, published,
quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006 34:
D689-D691.
For any further information or to express interest, please contact Nicolas Le Novère
(n.lenovere (at) gmail.com)
Proteins often have small absolute free energies of folding (ΔGfold)
and are frequently marginally stable at room temperature – the power of this on
biological systems is really familiar in the adaptive response of fever to an
infection – the body raises it’s temperature a few degrees with the aim of denaturing pathogen proteins and exposing antigens, slowing down replication, etc.. Raising a
human’s body temperature just a few degrees further is fatal (normal body temperature is ca. 37 C, temperatures above 40 C can be fatal). Similarly, lowering temperature a few degrees has serious effects on the ability for proteins
to do their job, and subsequently on normal everyday life. So proteins are only
required to be functional over a comparatively narrow range of operational
temperatures, and where temperature differences occur there can be evolutionary selection for this
trait (as an interesting example, sequencing studies have identified alleles in cod under selective pressure for temperature (link here)). This dependence on temperature is probably an extra significant factor
for proteins with two (or more) conformations for which the distribution/equilibrium is small anyway and is then ‘biased’ by small molecule binding.
An important
corollary to this is that ΔGfold at lab/body temperature is not necessarily
reflected in the Tm for a protein.
Some organisms regulate their body temperature (like me),
others don’t (like Vini and Bruce my Pogona) and just have to exist at
ambient temperatures – again typically there is a range over which normal
everyday life is possible, too cold and ‘cold-blooded’ life becomes sluggish
and dormant, too hot and they typically seek shelter and again become inactive.
This temperature dependence on function is implicit in
almost all of biology, and is so obvious that it is often unspoken what temperature
an assay is performed at – but temperature differences can easily give rise to
differences in bioassay results – especially for enzymes where rates of
reactions are very sensitive to temperature changes. As an example of some of the issues - here is the protocol for a panel of assays (just chosen at random) using the ubiquitous 'room temperature' which can be between 15 and 25 C). I guess all the controls have been done though, and in this case it doesn't matter.
One of the impacts that non-synonymous genetic mutations can cause are changes in protein stability, and in the limit a significantly destabilising coding
mutation will lead to completely non-functional protein - for example, by the introduction of a
charged residue in the hydrophobic core of a protein. Since this has the
physical appearance of a mis-folded protein it will be recognised as such and
tagged for clearance – so a double whammy - what minimal function remains is
itself rapidly cleared. However, we know that the vast majority of amino-acid
mutations are functionally neutral, which is fortunate for all of us.
This "temperature dependence of function" feature is not
regularly explored in drug discovery, but can take one some interesting places if you let your mind drift across the literature – so for example it leads to the hypothesis that it may be possible to
stabilise a destabilised protein with a small molecule – and this has been
tested a number of times – the best example that springs to mind is for p53 a key tumour suppressor in cancer cell biology – where screening at different temperatures has been used to identify small molecule stabilisers of mutant p53. Importantly this paper showed it was possible to find compounds (e.g. CP-31398) that perturbed a mutant protein back towards it's native state.
p53 stabiliser CP-31398
A great example of how normal function can be restored to a mutant protein is with the recently launched breakthrough drug for Cystic Fibrosis - Ivacaftor (VX-770, tradename Kalydeco).
Ivacaftor
As one step further, you could imagine performing an in vivo screen where you look for a
compound that narrows or perturbs the range at which an organism can effectively live by making it more temperature sensitive. Compounds with these properties could have great commercial application in various pest species.
Understanding the impact on thermodynamics and dynamics of 'unstable'/temperature-sensitive systems such as these is a great opportunity and is currently an under-explored area for future drug design. If you are interested in a PhD studentship in this area, please get in touch, and see this page for application details.
Following the scientific literature is a difficult task, it's not something you can do lying on the beach. Among the huge number of articles that are published every day only a few really matter to you. These are the articles you are going to have the time to read and that are relevant to your research.
In order to spot the relevant papers, there are a number of thematic approaches. For example, most journals publish articles in a relatively restricted field - and so a scientist can follow a thematic journal (like PLOS, Nature Biotechnology, etc). But then you'll still receive a lot of information to be sorted and you will obviously notice only the recent work. Maybe that interesting article you are waiting for has been published 5 years ago and you will then never ever notice it that way.
You can also keep track of literature with a keywords search (that you save somehow). This approach presents some problems too: What keywords should you search for? Your research is certainly more complex and subtle than "cancer p53" or "sequence analysis".
PubMed Watcher is an experimental tool designed to help biomedical scientists to read efficiently adequate literature. The tool is built based on the following assumptions:
Relevant papers are forever: PubMed Watcher abstracts away from the publishing date.
Relevant papers are everywhere: The tool considers all the articles indexed by PubMed.
Your research cannot be defined by a few keywords only: PubMed Watcher relies on Key Articles as a means to express the science you care about.
More details and the methodology behind the tool is presented here alongside an example to illustrate how it works.
PubMed Watcher is still in beta testing, please submit your feedback or idea for improvements here or directly by email.


 There are more and more software libraries being ported to JavaScript. The best example is JavaScript/HTML5 Citadel demo of the Unreal Engine. So why not to try with some chemical stuff? One of the most important chemical software libraries is IUPAC InChi. It's also extremely hard to reimplement as it's written in low-level, functional-style C. On the other hand it's just a few headers and source files, without any dependencies so it's a perfect use case for Emscripten.Emscripten 'is an LLVM-to-JavaScript compiler'. It can be used as a drop-in replacement for standard tools such as gcc or make. Recently it got support for
There are more and more software libraries being ported to JavaScript. The best example is JavaScript/HTML5 Citadel demo of the Unreal Engine. So why not to try with some chemical stuff? One of the most important chemical software libraries is IUPAC InChi. It's also extremely hard to reimplement as it's written in low-level, functional-style C. On the other hand it's just a few headers and source files, without any dependencies so it's a perfect use case for Emscripten.Emscripten 'is an LLVM-to-JavaScript compiler'. It can be used as a drop-in replacement for standard tools such as gcc or make. Recently it got support for Don’t we just love the fact that these days so much bioactivity data is freely available at no cost (to the end user)? I think we do. The more, the better. So, what would your answer be if someone asked you if you consider it to be a good idea if they would deposit some of their unpublished bioactivity data in ChEMBL? My guess is that you would be all in favour of this idea. 'Go for it', you might even say. On the other hand, if the same person would ask you what you think of the idea to deposit some of ‘your bioactivity data’ in ChEMBL the situation might be completely different.First and foremost you might respond that there is no such bioactivity data that you could share. Well let’s see about that later. What other barriers are there? If we cut to the chase then there is one consideration that (at least in my experience) comes up regularly and this is the question: 'What’s in it for me?' Did you ask yourself the same question? If you did and you were thinking about ‘instant gratification’ I haven’t got a lot to offer. Sorry, to disappoint you. However, since when is science about ‘instant gratification’? If we would all start to share the bioactivity data that we can share (and yes, there is data that we can share but don’t) instead of keeping it locked up in our databases or spreadsheets this would make a huge difference to all of us. So far the main and almost exclusive way of sharing bioactivity data is through publications but this is (at least in my view) far too limited. In order to start to change this (at least a little bit) the concept of ChEMBL supplementary bioactivity data has been introduced (as part of the efforts of the Open PHACTS project, http://www.openphacts.org).Here is how it works: If you have unpublished bioactivity data that has been generated in an assay that can be found in ChEMBL (since the publication where the assay is described is also in ChEMBL), you can now deposit this data in ChEMBL (see http://dx.doi.org/10.6019/CHEMBL2094195 for an example). The obvious situation would be one where only a subset of the results have been reported in the publication but there are many more results (e.g. inactives). If you work in an industrial setting and might feel that you are not be in a position to release additional chemical structures you could think about depositing bioactivity data for compounds in (older) patents. Or you have reported bioactivity data in a poster. These are only examples and there are many more opportunities. In some cases we might explore new territory and the progress might be slow, but if we don’t try new things we are stuck with what we have. 'Do we really want this?' I hope the answer is no. So, let’s not focus on ‘instant gratification’ but help to grow the body of freely available bioactivity data by contributing to ChEMBL supplementary bioactivity data. If we could just give it a go it might make a difference. The concept might be quite restricted (e.g. the assay needs to be published) but we need to start somewhere. If you want to find out more about ChEMBL supplementary bioactivity data why not drop ChEMBL Help a line (chembl-help@ebi.ac.uk) and put ‘ChEMBL supplementary bioactivity data’ in the subject field. And don’t worry, you are not committing yourself by wanting to know more.ChEMBL, and the whole world of drug discoverers, is looking forward to hearing from you.Stefan Senger
Don’t we just love the fact that these days so much bioactivity data is freely available at no cost (to the end user)? I think we do. The more, the better. So, what would your answer be if someone asked you if you consider it to be a good idea if they would deposit some of their unpublished bioactivity data in ChEMBL? My guess is that you would be all in favour of this idea. 'Go for it', you might even say. On the other hand, if the same person would ask you what you think of the idea to deposit some of ‘your bioactivity data’ in ChEMBL the situation might be completely different.First and foremost you might respond that there is no such bioactivity data that you could share. Well let’s see about that later. What other barriers are there? If we cut to the chase then there is one consideration that (at least in my experience) comes up regularly and this is the question: 'What’s in it for me?' Did you ask yourself the same question? If you did and you were thinking about ‘instant gratification’ I haven’t got a lot to offer. Sorry, to disappoint you. However, since when is science about ‘instant gratification’? If we would all start to share the bioactivity data that we can share (and yes, there is data that we can share but don’t) instead of keeping it locked up in our databases or spreadsheets this would make a huge difference to all of us. So far the main and almost exclusive way of sharing bioactivity data is through publications but this is (at least in my view) far too limited. In order to start to change this (at least a little bit) the concept of ChEMBL supplementary bioactivity data has been introduced (as part of the efforts of the Open PHACTS project, http://www.openphacts.org).Here is how it works: If you have unpublished bioactivity data that has been generated in an assay that can be found in ChEMBL (since the publication where the assay is described is also in ChEMBL), you can now deposit this data in ChEMBL (see http://dx.doi.org/10.6019/CHEMBL2094195 for an example). The obvious situation would be one where only a subset of the results have been reported in the publication but there are many more results (e.g. inactives). If you work in an industrial setting and might feel that you are not be in a position to release additional chemical structures you could think about depositing bioactivity data for compounds in (older) patents. Or you have reported bioactivity data in a poster. These are only examples and there are many more opportunities. In some cases we might explore new territory and the progress might be slow, but if we don’t try new things we are stuck with what we have. 'Do we really want this?' I hope the answer is no. So, let’s not focus on ‘instant gratification’ but help to grow the body of freely available bioactivity data by contributing to ChEMBL supplementary bioactivity data. If we could just give it a go it might make a difference. The concept might be quite restricted (e.g. the assay needs to be published) but we need to start somewhere. If you want to find out more about ChEMBL supplementary bioactivity data why not drop ChEMBL Help a line (chembl-help@ebi.ac.uk) and put ‘ChEMBL supplementary bioactivity data’ in the subject field. And don’t worry, you are not committing yourself by wanting to know more.ChEMBL, and the whole world of drug discoverers, is looking forward to hearing from you.Stefan Senger

 A great example of how normal function can be restored to a mutant protein is with the recently launched breakthrough drug for Cystic Fibrosis - Ivacaftor (VX-770, tradename Kalydeco).
A great example of how normal function can be restored to a mutant protein is with the recently launched breakthrough drug for Cystic Fibrosis - Ivacaftor (VX-770, tradename Kalydeco). As one step further, you could imagine performing an in vivo screen where you look for a compound that narrows or perturbs the range at which an organism can effectively live by making it more temperature sensitive. Compounds with these properties could have great commercial application in various pest species.
As one step further, you could imagine performing an in vivo screen where you look for a compound that narrows or perturbs the range at which an organism can effectively live by making it more temperature sensitive. Compounds with these properties could have great commercial application in various pest species.




