-
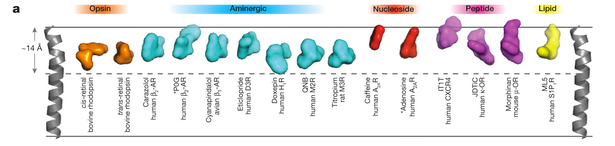
Paper: Molecular Signatures of G-protein-coupled Receptors

There's a nice review (shame about some of the proof-reading though!) of GPCR structure-function in this weeks Nature, it brings together a current overview of the known structures, their comparison, binding sites, and is a great introduction to the field.
Link to the paper here.
%A A.J. Vankatakrishnan %A X. Deupi %A G. Lebon %A C.G. Tate %A G.F. Schertler %A M.M. Babu %T Molecular Signatures of G-Protein-coupled Receptors %J Nature %V 494 %P 183-194 %D 2013 %O doi.10.1038/nature11896
-
Latest activities on the Activities table in ChEMBL_15
 For the recent ChEMBL_15 release, a considerable part of our efforts was focussed on the standardisation and harmonisation of the data in the Activities table. The latter holds all the quantitative and qualitative experimental measurements across compounds, assays and targets; needless to say that without it there's no ChEMBL!This is a summary of what we've incorporated so far:
For the recent ChEMBL_15 release, a considerable part of our efforts was focussed on the standardisation and harmonisation of the data in the Activities table. The latter holds all the quantitative and qualitative experimental measurements across compounds, assays and targets; needless to say that without it there's no ChEMBL!This is a summary of what we've incorporated so far:- Flag missing data: Records with null published values and null activity comments were flagged as missing.
- Standardise activity types and units: Conversion of heterogenous published activity type descriptions and units to a standard_type and set of standard_units (e.g., for IC50 convert mM/uM/pM measurements to nM).
- Flag unusual units: Records with unusual published units for their respective activity types were flagged as 'non standard'. For example, a hypothetical record with IC50 type and units in kg would be flagged!
- Convert the log values: The records with activity types such as pKi and logIC50 were appropriately converted to their non-log equivalents (by considering the units and sign of course as well). This updated a whopping 25% of the activities table - this means that significantly more data will become more comparable for subsequent analyses.
- Round values: For records with a standard activity value above 10, the rounding was done to the second decimal place. Otherwise, rounding was performed after the first three significant digits. For example 0.00023666666 would become a more concise 0.000237
- Check activity ranges: Records with a standard activity value outside the range specified by our expert biological curators, given the standard unit and type, were appropriately flagged.
- Detect duplicated values: For this one, we were inspired by a recent publication. What we did is we detected and flagged duplicated activity entries and potential transcription errors in activity records that come from publications. The former are records with identical compound, target, activity, type and unit values that were most likely reported as citations of measurements from previous papers, even when these measurements were subsequently rounded. The latter cases consist of otherwise identical entries whose activity values differ by exactly 3 or 6 orders of magnitude indicating a likely error in the units (e.g. uM instead of nM).
As a result of our efforts, we added 2 new columns in the Activities table, namely Data_validity_comment and Potential_duplicate. The former takes one out of 5 possible values: NULL, 'Potential missing data' (see point 1), 'Non standard unit for type' (see point 3), 'Outside typical range' (see point 6) and 'Potential transcription error' (see point 7). The latter column contains a binary (0,1) flag to indicate whether we think the specific activity record is a duplicate, as per point 7 above.Stay tuned for more posts on the changes/improvements introduced by the new ChEMBL_15 release. Meanwhile, if you have any comments/feedback on the curation process or on the activity types we should prioritise, please let us know!
George -
New Drug Approvals 2013 - Pt. II - Mipomersen (KynamroTM)

On January 29st, the FDA approved Mipomersen (Tradename: Kynamro; Research Code: ISIS-310312), an oligonucleotide inhibitor of apolipoprotein B-100 (apo B-100) synthesis, indicated as an adjunct to lipid-lowering medications and diet to reduce low density lipoprotein-cholesterol (LDL-C), apolipoprotein B (apo B), total cholesterol (TC), and non-high density lipoprotein-cholesterol (non HDL-C) in patients with homozygous familial hypercholesterolemia (HoFH).
Familial hypercholesterolemia is a genetic disorder, characterised by high levels of cholesterol rich low-density lipoproteins (LDL-C) in the blood. This genetic condition is generally attributed to a faulty mutation in the LDL receptor (LDLR) gene, which mediates the endocytosis of LDL-C.
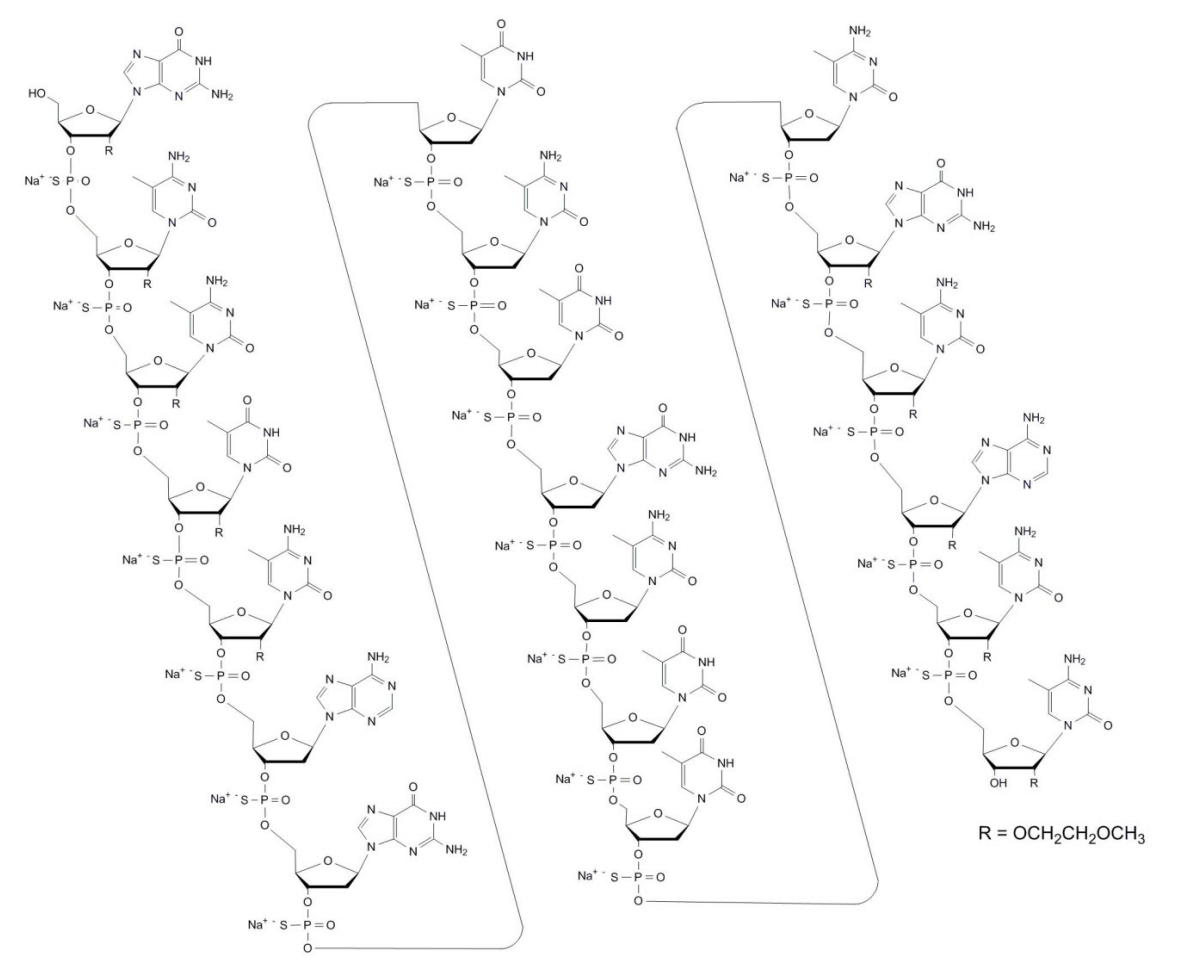
Mipomersen is the first antisense oligonucleotide that targets messenger RNA (mRNA) enconding apolipoprotein B-100 (Apo B-100), the principal apolipoprotein of LDL and its metabolic precursor, very low density lipoprotein (VLDL). Mipomersen forms a duplex with the target mRNA, causing the mRNA to be cleaved by RNase H and therefore unable to be translated to apoB-100. Hepatic apoB mRNA silencing gives rise to reductions in hepatic apoB, total cholesterol and LDL-C levels in the serum (PMID: 23226021).
The binding site for mipomersen lies within the coding region of the apo B mRNA at the position 3249-3268 relative to the published sequence GenBank accession number NM_000384.1.
Mipomersen, like other antisense oligonucleotides, belongs to the -rsen USAN/INN stem group. Several members of this class are currently in clinical trials like Alicaforsen (Isis, phase III) for the treatment of inflammatory bowel disease and Oblimersen (Genta, phase II) for cancer therapy.
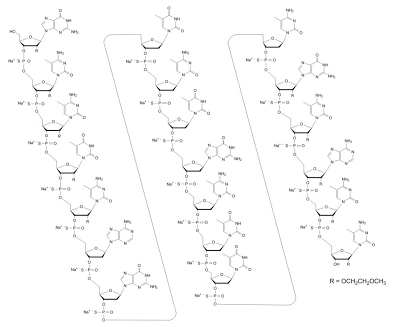
Mipomersen (IUPAC Name: 2'-O-(2-methoxyethyl)-P-thioguanylyl-(3'→5')-2'-O-(2-methoxyethyl)-5-methyl- P-thiocytidylyl-(3'→5')-2'-O-(2-methoxyethyl)-5-methyl-P-thiocytidylyl-(3'→5')- 2'-O-(2-methoxyethyl)-5-methyl-P-thiouridylyl-(3'→5')-2'-O-(2-methoxyethyl)-5- methyl-P-thiocytidylyl-(3'→5')-2'-deoxy-P-thioadenylyl-(3'→5')-2'-deoxy-P- thioguanylyl-(3'→5')P-thiothymidylyl-(3'→5')-2'-deoxy-5-methyl-P-thiocytidylyl- (3'→5')-P-thiothymidylyl-(3'→5')-2'-deoxy-P-thioguanylyl-(3'→5')-2'-deoxy-5- methyl-P-thiocytidylyl-(3'→5')-P-thiothymidylyl-(3'→5')-P-thiothymidylyl- (3'→5')-2'-deoxy-5-methyl-P-thiocytidylyl-(3'→5')-2'-O-(2-methoxyethyl)-P- thioguanylyl-(3'→5')-2'-O-(2-methoxyethyl)-5-methyl-P-thiocytidylyl-(3'→5')-2'- O-(2-methoxyethyl)-P-thioadenylyl-(3'→5')-2'-O-(2-methoxyethyl)-5-methyl-P- thiocytidylyl-(3'→5')-2'-O-(2-methoxyethyl)-5-methylcytidine nonadecasodium salt; Canonical smiles: COCCO[C@H]1[C@@H](O)[C@H](COP(=O)(O)S[C@H]2[C@H](COP(=O)(O)S[C@H]3[C@H](COP(=O)(O)S[C@H]4[C@H](COP(=O)(O)S[C@@H]5[C@@H](COP(=O)(O)S[C@H]6C[C@@H](O[C@@H]6COP(=O)(O)S[C@H]7C[C@@H](O[C@@H]7COP(=O)(O)S[C@H]8C[C@@H](O[C@@H]8COP(=O)(O)S[C@H]9C[C@@H](O[C@@H]9COP(=O)(O)S[C@H]%10C[C@@H](O[C@@H]%10COP(=O)(O)S[C@H]%11C[C@@H](O[C@@H]%11COP(=O)(O)S[C@H]%12C[C@@H](O[C@@H]%12COP(=O)(O)S[C@H]%13C[C@@H](O[C@@H]%13COP(=O)(O)S[C@H]%14C[C@@H](O[C@@H]%14COP(=O)(O)S[C@H]%15C[C@@H](O[C@@H]%15COP(=O)(O)S[C@@H]%16[C@@H](COP(=O)(O)S[C@@H]%17[C@@H](COP(=O)(O)S[C@@H]%18[C@@H](COP(=O)(O)S[C@@H]%19[C@@H](COP(=O)(O)S[C@@H]%20[C@@H](CO)O[C@H]([C@@H]%20OCCOC)n%21cnc%22C(=O)NC(=Nc%21%22)N)O[C@H]([C@@H]%19OCCOC)N%23C=C(C)C(=NC%23=O)N)O[C@H]([C@@H]%18OCCOC)N%24C=C(C)C(=NC%24=O)N)O[C@H]([C@@H]%17OCCOC)N%25C=C(C)C(=O)NC%25=O)O[C@H]([C@@H]%16OCCOC)N%26C=C(C)C(=NC%26=O)N)n%27cnc%28c(N)ncnc%27%28)n%29cnc%30C(=O)NC(=Nc%29%30)N)N%31C=C(C)C(=O)NC%31=O)N%32C=C(C)C(=NC%32=O)N)N%33C=C(C)C(=O)NC%33=O)n%34cnc%35C(=O)NC(=Nc%34%35)N)N%36C=C(C)C(=NC%36=O)N)N%37C=C(C)C(=O)NC%37=O)N%38C=C(C)C(=O)NC%38=O)N%39C=C(C)C(=NC%39=O)N)O[C@H]([C@@H]5OCCOC)n%40cnc%41C(=O)NC(=Nc%40%41)N)O[C@@H]([C@H]4OCCOC)N%42C=C(C)C(=NC%42=O)N)O[C@@H]([C@H]3OCCOC)n%43cnc%44c(N)ncnc%43%44)O[C@@H]([C@H]2OCCOC)N%45C=C(C)C(=NC%45=O)N)O[C@@H]1N%46C=C(C)C(=NC%46=O)N; ChEMBL: CHEMBL1208153; Standard InChI Key: TZRFSLHOCZEXCC-PBNBMMCMSA-N) is a synthetic second-generation 20-base phosphorothioate antisense oligonucleotide, with a molecular weight of 7594.9 Da and the following sequence:
5'-GMeCMeCMeUMeCAGTMeCTGMeCTTMeCGMeCAMeCMeC-3'
where the underlined residues are 2′-O-(2-methoxyethyl) nucleosides, and the remaining are 2′-deoxynucleosides. Substitution at the 5-position of the cytosine (C) and uracil (U) bases with a methyl group is indicated by Me.
Mipomersen is available as an aqueous solution for subcutaneous injection, and the recommended weekly dose is a single-use pre-filled syringe containing 1 mL of a 200 mg/mL solution. Following subcutaneous injection, peak concentrations of mipomersen are typically reached in 3 to 4 hours. The estimated plasma bioavailability of mipomersen following subcutaneous administration over a dose range of 50 mg to 400 mg, relative to intravenous administration, ranged from 54% to 78%. Mipomersen is highly bound to human plasma proteins (≥ 90%).
Mipomersen is metabolised in tissues by endonucleases to form shorter oligonucleotides that are then substrates for additional metabolism by exonucleases, and finally excreted in urine. The elimination half-life (t1/2) for mipomersen is approximately 1 to 2 months.
Mipomersen has been approved with a black box warning due to an increase in transaminases (alanine aminotransferase [ALT] and/or aspartate aminotransferase [AST]) levels, and hepatic fat content (steatosis) after exposure to the drug.
The license holder for KynamroTM is Genzyme Corporation, and the full prescribing information can be found here. -
Future Webinars

After the success of the last round of webinars, we have decided to run another set.
However, we would like to gauge the interest in which topics would be most useful. The topics that have been suggested so far are:
- ChEMBL Overview - Basic interface walkthrough and searching
- ChEMBL Schema - Basic overview and ChEMBL changes
- ChEMBL Schema - Changes to ChEMBL target data model
- UniChem - Basic overview and searching
- Drug and USAN data content
For this, a doodle poll has been set up which will allow you to register your interest. The poll is hidden, so no one will see your details, but it will allow us at ChEMBL Towers to see if the webinar would be worthwhile to set up. Please click on the link and let us know what you think.
Additionally, if there are any webinars that we have not suggested, that you believe would be useful, please feel free to suggest these on the doodle poll, or email chembl-help@ebi.ac.uk -
New Drug Approvals 2013 - Pt. 1 - Alogliptin (NesinaTM)
On January 25th 2013, FDA approved Alogliptin (as the benzoate salt; tradename: Nesina; research code: SYR-322, TAK-322; CHEMBL: CHEMBL376359), a dipeptidyl peptidase-4 (DPP-4) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus (also known as noninsulin-dependent diabetes mellitus (NIDDM)).
NIDDM is a chronic disease characterized by high blood glucose. In response to meals, increased concentrations of incretin hormones such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are released into the bloodstream from the small intestine. These hormones cause insulin release from the pancreatic beta cells in a glucose-dependent manner but are inactivated by the DPP-4 enzyme within minutes. GLP-1 also lowers glucagon secretion from pancreatic alpha cells, reducing hepatic glucose production. In patients with NIDDM, concentrations of GLP-1 are reduced but the insulin response to GLP-1 is preserved. Alogliptin exerts its therapeutic action by inhibiting DPP-4, thereby slowing the inactivation of the incretin hormones and increasing their bloodstream concentrations.
Image from Wikipedia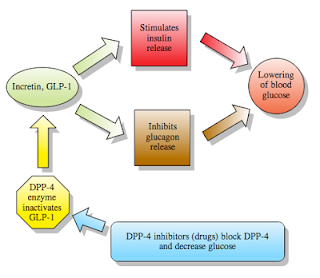
Other DPP-4 inhibitors are already available on the market (some of which have already been covered here on the ChEMBL-og) and these include Linagliptin (approved in 2011 under the tradename Tradjenta; ChEMBL: CHEMBL237500; PubChem: CID10096344; ChemSpider: 8271879), Saxagliptin (approved in 2009 under the tradename Onglyza; ChEMBL: CHEMBL385517; PubChem: CID11243969; ChemSpider: 9419005) and Sitagliptin (approved in 2006 under the tradename Januvia; ChEMBL: CHEMBL1422; PubChem: CID4369359; ChemSpider: 3571948). Several other DPP-4 inhibitors are in clinical trials such as Trelagliptin (ChEMBL: CHEMBL1650443; research code: SYR-472), Omarigliptin (ChEMBL: CHEMBL2105762; research code: MK-3102), Carmegliptin (ChEMBL: CHEMBL591118; research code: R-1579, RO-4876904), Gosogliptin (ChEMBL: CHEMBL515387; research code: PF-734200), Dutogliptin (research code: PHX1149), Denagliptin (ChEMBL: CHEMBL2110666; research code: GW823093). Vildagliptin (ChEMBL: CHEMBL142703) has been approved in Europe and Japan, but not in the United States.
DPP-4 (ChEMBL: CHEMBL284; Uniprot: P27487) is 766 amino acid-long enzyme, which is responsible for the removal of N-terminal dipeptides sequentially from polypeptides having unsubstituted N termini, provided that the penultimate residue is proline. It belongs to the Dipeptidyl peptidase IV family (PFAM: PF00930).
>DPP4_HUMAN Dipeptidyl peptidase 4 MKTPWKVLLGLLGAAALVTIITVPVVLLNKGTDDATADSRKTYTLTDYLKNTYRLKLYSL RWISDHEYLYKQENNILVFNAEYGNSSVFLENSTFDEFGHSINDYSISPDGQFILLEYNY VKQWRHSYTASYDIYDLNKRQLITEERIPNNTQWVTWSPVGHKLAYVWNNDIYVKIEPNL PSYRITWTGKEDIIYNGITDWVYEEEVFSAYSALWWSPNGTFLAYAQFNDTEVPLIEYSF YSDESLQYPKTVRVPYPKAGAVNPTVKFFVVNTDSLSSVTNATSIQITAPASMLIGDHYL CDVTWATQERISLQWLRRIQNYSVMDICDYDESSGRWNCLVARQHIEMSTTGWVGRFRPS EPHFTLDGNSFYKIISNEEGYRHICYFQIDKKDCTFITKGTWEVIGIEALTSDYLYYISN EYKGMPGGRNLYKIQLSDYTKVTCLSCELNPERCQYYSVSFSKEAKYYQLRCSGPGLPLY TLHSSVNDKGLRVLEDNSALDKMLQNVQMPSKKLDFIILNETKFWYQMILPPHFDKSKKY PLLLDVYAGPCSQKADTVFRLNWATYLASTENIIVASFDGRGSGYQGDKIMHAINRRLGT FEVEDQIEAARQFSKMGFVDNKRIAIWGWSYGGYVTSMVLGSGSGVFKCGIAVAPVSRWE YYDSVYTERYMGLPTPEDNLDHYRNSTVMSRAENFKQVEYLLIHGTADDNVHFQQSAQIS KALVDVGVDFQAMWYTDEDHGIASSTAHQHIYTHMSHFIKQCFSLP
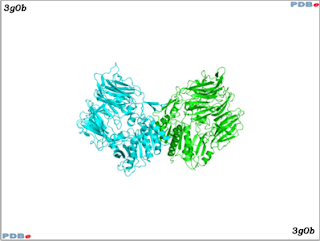
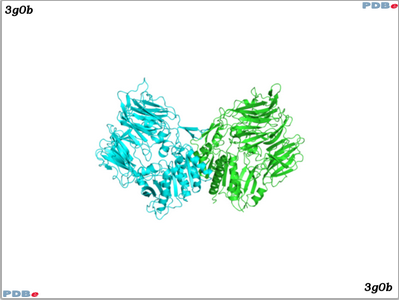
The image above shows a crystal structure of DPP-4 (in this example, two copies of DPP-4 are displayed - PDBe: 3g0b). Information on the active site of DPP-4 can be found here.


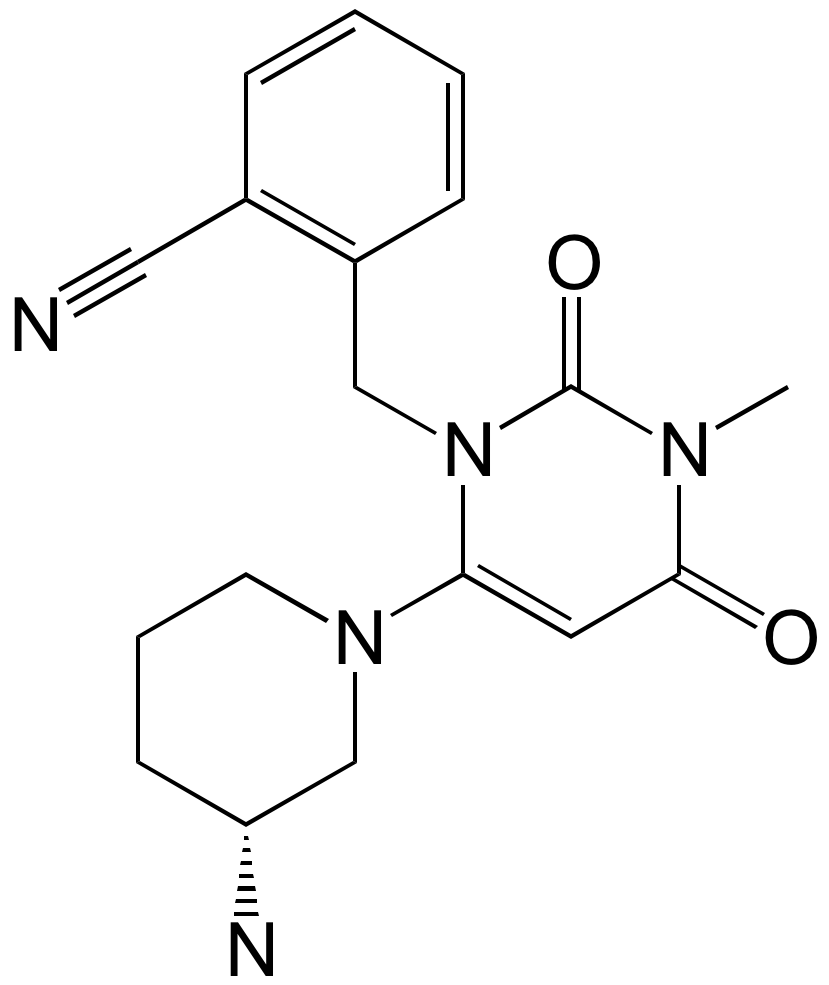
Alogliptin is an oral small-molecule with a molecular weight of 339.4 Da (461.51 Da as the benzoate salt). The image on the right shows Alogliptin in the active site of DPP-4. Important features of its chemical structure are the aminopiperidine motif, which provides a salt bridge to the glutamic acids residues 205/206 in the active site of DPP-4, the cyanobenzyl group which interacts with the arginine residue 125, the carbonyl group from the pyrimidinedione moiety which participates in an hydrogen bond with the backbone NH of tyrosine 631 and the uracil ring which π-stacks with tyrosine 547.
IUPAC: 2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxopyrimidin-1-yl]methyl]benzonitrile
Canonical Smiles: CN1C(=O)C=C(N2CCC[C@@H](N)C2)N(Cc3ccccc3C#N)C1=O
InChI: InChI=1S/C18H21N5O2/c1-21-17(24)9-16(22-8-4-7-15(20)12-22)23(18(21)25)11-14-6-3-2-5-13(14)10-19/h2-3,5-6,9,15H,4,7-8,11-12,20H2,1H3/t15-/m1/s1
The recommended dosage of Alogliptin is 25 mg once daily. Alogliptin has good oral bioavailability F (approximately 100% bioavailable), with a volume of distribution Vd of 417 L and a low plasma protein binding (20%). Excretion is mainly renal (76% of the dose recovered in urine) and mostly as the parent compound (60% to 71%). Alogliptin is metabolized by CYP2D6 and CYP3A4 to two minor metabolites, M-I (N-demethylated alogliptin - >1% of the parent drug), which is an active metabolite and is an inhibitor of DPP-4 similar to the parent molecule and M-II (N-acethylated alogliptin - >6% of the parent drug), which does not display any inhibitory activity towards DPP-4 or other DPP-related enzymes. The renal clearance of Alogliptin is 9.6 L/hr and the systemic clearance is 14.0 L/hr.
The license holder is Takeda Pharmaceuticals America, Inc. and the prescribing information of Alogliptin can be found here.
Patricia -
Japan - Here I Come (in October)!

I'm out in Japan at the end of October this year - the week of October 28th 2013 for a scientific conference (the CBI Annual Conference). Japan is one of my favorite places in the whole world, and I have a routine of...
- browsing vintage hi-fi shops
- hunting out high-end capacitor and choke components,
- eating eel うなぎ bento,
- visiting CD stores (at least the format lives on in Japan, and the Obi Strips and enhanced content makes compelling browsing for an obsessive completist like me)
- going to Bic Camera 株式会社ビックカメラ
- visiting Akihabara 秋葉原.
My schedule is currently empty for Wednesday 30th and Thursday 31st. I'd be delighted to visit and give a seminar, or maybe run a workshop on ChEMBL, so if you are interested in meeting up, or a visit, or an evening meal, let me know.
jpo -
The Ontogeny and Evolution of Compound Names
Here's a post prompted by a wipe board discussion with a visitor the other day - they asked if I had ever written this down, and so here it is.
The naming of compounds is one of the most enabling and frustrating aspects of chemoinformatics - and getting a handle on compound naming conventions is one of the big gotcha's for people entering the field from bioinformatics, where things are frankly a lot simpler (yes, I know it's not easy there - but it's a lot easier than in bioinformatics).
It's interesting to view the naming of compounds in a timeline manner, and for those of you with a software bent, in the context of variable names....
I describe here a prototypical typical system - electronic lab-books and so forth will of course change the details. When a chemist makes a compound they know the 2-D structure (or they will work it out once they purify the product) and it has a lab-book reference. This could be something like 2341/23/4 (which could encode that the particular compound is labelled as compound 4 on page 23 of a lab book numbered 2341), once the compound is purified and registered in a compound database at an institute it will typically get a research code - something like EBI-7821-1 - there is usually some convention here to the naming - EBI means it was made at the European Bioinformatics Institute (this is a made up example, we don't make compounds!), and it is the 7,821th compound made, and then the -1 means that it is batch 1 - since when we make it again, it is likely to have a different purity, contaminants, and potentially differing biological activity. These are all private names, the world doesn't see any of these.
Then a chemist writes up the work, and publishes it in a journal - for brevity (and other reasons) compounds are typically published with a paper identifier like 12d within a particular paper - it is just a tag within a particular paper, with the scope of the name limited to a particular publication - just like a local variable in a software function. There will be many other completely unrelated compounds spread across the literature - this type of name is not useful for integration purposes.
If you write a patent on the compound, it will typically be named with it's IUPAC name, and/or with a patent identifier number - e.g. example 27, again a local variable within a patent. There will be no relationship between the patent local name and the publication identifier. These are public names.
So why don't people more generally use IUPAC names - well they are often long, difficult to say and pronounce without sounding like a fool, difficult to remember, difficult to type, and there are often many valid alternate IUPAC names for a particular compound - so they are usually avoided completely when talking about a compound. CAS numbers are a further alternative, the issue there is that a private organisation holds the naming rights, they are not public, there are tights restrictions on how many you can store, and using them is very expensive.
So far we have gone from a lab book reference (private), to a research code (private), to a local identifier (public) in a paper/patent, and for all of these we have a 2-D structure.
If the compound is sufficiently interesting and more people use it, and it is published in other publications, there comes a point when the research code is published and the therefore becomes public. Occasionally this happens on first publication, but it's pretty unusual.
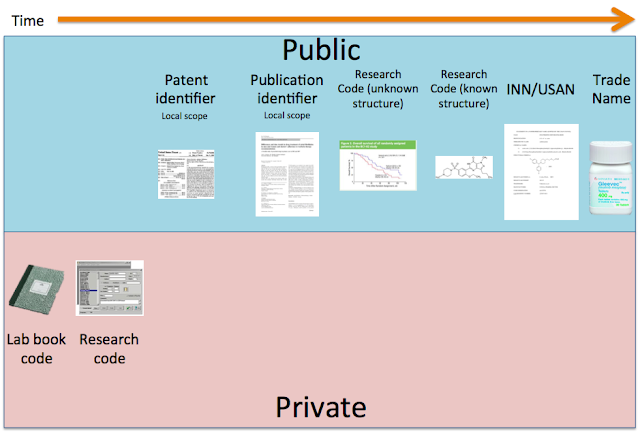
More usual though is that a research code will appear in the literature with some interesting biological properties - something like EBI-9437, which imagine - makes stem cells differentiate into competent beta cells. The batch is not usually reported (and referees would rightly be sniffy about batch specific effects, and by the stage that people report these results the synthesis, purification and assay variability will be sorted out). It is probably the case that the structure of EBI-9437 is already published, in a patent usually - it's just that you don't know what particular compound out of many hundreds that it actually is. So for some of the most interesting recent compounds we know their research code (and usually where they come from from the syntax of the name) but not the structure. Many people outside the organization that made EBI-9437 would like to know the structure.
At some point, usually at a big meeting or in a high profile publication, the structure of EBI-9437 will be shown to the world. At this point, it can be mapped back to all the previous names (on the basis of 2D structure).
If the compound is commercially interesting, it could be licensed to another company, who often want to make it clear to the world that they control the compound, or to imply that it came from their own labs, so they may rename it. So in this case imagine our EBI-9437 becomes WTF-433932, after the rights to the compound are acquired by WTF Discovery Inc.
Many compounds stop here in terms of their progression of names - they may be too toxic for wide use, they may just not be enough money to develop them further, or they may have just been tried in the wrong assay. For the 'luckier' compounds they will typically progress to full clinical development, still using their research codes, and typically scientific documents submitted to regulators (as part of and IND or review package) use Research Codes - many of these documents are in the public domain.
When a compound gets to this stage, compound vendors offer for the compound for sale, often assigning their own catalog numbers, these are easily confused with research codes, and it is an unfortunate feature that these are starting to contaminate both public chemical databases, and to some extent the literature; with this variable scope view these are really just local variables applicable over a particular compound suppliers catalog.
For those that look good in development, and there is the potential that the compound will be used in a product, a formal non-proprietary name is assigned - nowadays there are two major names USAN, and INN (both of these are public) - I've discussed these in the past, both the plusses and the minuses in their respective systems, so I won't discuss them here further - do a search on the blog for some more pointers if you are interested. In our case, for the fictional WTF-433932, imagine it's a tyrosine kinase inhibitor and gets assigned the name faybelitinib. At this point, further discussion and publications on the compounds should use these public USAN names. This is a great help, it is a single unambiguous name for a compound, but typically comes quite late in the lifecycle of a compound - and to get more information on the properties of a compound with a USAN name, you need to know the 2D structure, Research Code, patent ids, paper ids, etc. It is interesting that with the development of their own independent pharmaceutical discovery and development capabilities, nations such as China are assigning their own non-proprietary names, following stem conventions. These are not currently easily accessible though.
Finally, a compound will get a Trade or Brand Name, this is often geographically local in scope (so a drug will have different names in different markets e.g. Glivec and Gleevec, or different names in the same geographical market for different diseases e.g. Revatio and Viagra). So, for scientific reasons these are not reliable useful names, and should, in general be avoided.
So what we have is a hierarchy of names on increasing scope and consistency as a compound progresses from round-bottomed flask to patient. Because of the conditional and temporal assignments of these names of ever increasing scope and formality, the tracking to previous names is an essential task. Suffice it to say that Research Codes are where the action is.
jpo -
Chemical data provenance and a big pile of mess that should be celebrated on National Mess Day
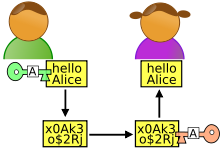
One of the increasing problems for those working in the chemical database field is one of distinguishing between primary and secondary sources, especially if salt stripping, normalization, errors, and assignment of new id's happen in this process. Here is a tale....
Alice is publicly spirited and puts her chemical database of 1000 compounds in the public domain. She assigns a clear permissive license to her data, and encourages reuse by providing a suitable download format. Her hope is that people, somewhere will see that she has these particular compounds, and they will contact her and collaborate Alice, maybe.
Bob has a similarly public spirit, and has 500 compounds to share. He has the same hope that people will search his database and collaborate with him.
Carlos is interested in compounds containing a pyridine group. So he wants to search both Alice's and Bob's databases - he could either search both Alice's and Bob's separate databases (assuming that they have the facilities in their labs to run a database system) - or he could use a system that Trent has built, Trent doesn't have any actual compounds, but he is a good programmer, and can integrate the data from Alice and Bob, so that Carlos doesn't need to do two searches, and then merge the results. Trent's system is liked by quite a few people, and so others start to contact Trent with their data. Trent contributes a lot to operation of the community, he is an independent broker of data, and simplifies the field by providing federated searches.
Alice and Bob are both busy people, and now some compounds they had are no longer available, Carlos found a few compounds, and so did a few of Carlos' friends, so these were sent in the post and disappeared from Alice and Bob's collections. However, Alice and Bob also made some new compounds and wanted to share these to, since maybe they would help Dan discover new drugs for trypanosomiasis. Alice and Bob update their local databases to reflect the deleted and new chemicals, and since they like to know a little about the history of the chemicals that they had, and need to write reports about how many new compounds they have made over the past year, they provide versions on their databases, since they can track when new things appeared. Trent should of course keep an eye on Alice's and Bob's sites and update his system appropriately.
Trent largely works on his own though, and doesn't have a lot of time, in fact his employer wasn't really keen on this work, but tolerated Trent's activities, and managed to justify it under another grant. Of course when Trent first planned his system he wrote it without thinking about future updates, it was just a fun hack - build it once, merge this cool chemical data and show the value. All that code to administer the federated database are just a drag, and the users never see all that effort.
Trent gets a new job in a different city, and he and his partner adopt a child, this means Trent has even less time to keep things in his database up to date.
Dan is getting increasingly frustrated though, since Trent's system is now out of date - although he relied on Trent having all of Alice's and Bob's data in one place, when he looks at it he sees it's old data, say five years out of date and Alice has got a whole bunch of new compounds that no-one seems interested in, because they use Trent's system.
Erin sees this problem and tries to sort it out; she then takes Trent's data, and decides to add Frank and George's data, which is new to her system. But relying on Trent's versions of Alice's and Bob's data (and Trent had assigned new identifiers as part of his merging of the compounds from Alice and Bob) has meant that she is serving old data, and no-one can possibly work out where on earth anything has actually come from.
Trent gets a new job again, and his kid goes to kindergarten, so he gets stuck back into this project, he decides to build on top of Erin's work and adds new data from Henry and Irma.
James doesn't know too much about chemoinformatics, he's a lab biologist just needing some ideas for compounds to test in an assay he's developed that models DNA methylation in Aplysia, and thinks that Trent's pages contain everything.
You'll agree that this is a fictional story - but how far divorced from reality is it? We ourselves face big problems when we try and take secondary sources (those that consolidate data from other 'primary' sources), and work out what is new data and what is processed old data. There is a clear subset of ChEMBL that is primary in the sense that we are the first point of entry on the data onto the internet, and we actively curate this "primary ChEMBL" set - but we certainly can't curate other people's data, and have this survive refreshes from the primary sources. We also make statements like "yeah, we have PubChem structures in ChEMBL", but of course we took a subset, applied some filters, and there is some delay in us taking a snapshot and processing it into ChEMBL. So if you want the latest, most definitive data from PubChem, go to PubChem.
Over this year, we will address the provenance of data that we load into UniChem and ChEMBL, making it easier for our users to know exactly what versions of what they are using. If others are interested in working with us on some community mechanism for versioning and persistence of parent source identifiers, let us know. Continuing the explosion of alternate secondary, tertiary, and so forth identifiers for compounds is not a good way forward, for anyone.
"Farm Fresh ChEMBLTM" is always available from us, there are a number of other places you can get fresh ChEMBL (a number of groups are already working hard on ChEMBL 15 integration into their systems). Some of the ChEMBLs out there aren't so fresh, so like every good consumer, check the "produced on" date!
The names have some some deeper semantic meaning here (look at the Alice and Bob wikipedia page - of course there are inevitably a few Chuck's around as well trying to lock data behind paywalls or search boxes).
jpo