The following images show the main changes (in this example, for the case of an oral synthetic small molecule):
1. We have visually separated the ingredient-specific information (icons in green) from the product-specific information (icons in blue).
2. The chirality icon will now also show if the ingredient is dosed as a racemic mixture (an image of two human hands).
3. An extra icon has been added to indicate the marketing status of a drug product. The product can be available as prescription (an image of the letters RX), over-the-counter (an image of the letters OTC) or discontinued (an image of the letters of RX with a stripe across it).
In summary...
The ingredient icons (in green) display the following information (from left to right)
Drug class
this can either be
Synthetic small molecule
Natural product-derived small molecule
Inorganic small molecule
Peptide/protein
Monoclonal antibody
Enzyme
Oligonucleotide
Oligosaccharide.
Rule of Five
An image of the number five: this is either pass or fail - we fail a molecule if it fails to pass all the individual tests (usually people use fail one parameter); we use AlogP for the calculations and use 5.0 as a cutoff.
New target
An image of a 'bullseye' target: this is either true or false - the target here refers to the molecular target responsible (or believed to be responsible) for its therapeutic efficacy.
Chirality
An image of two human hands: the drug is dosed as a racemic mixture.
An image of a chiral human hand: the drug is dosed as a single optically active substance.
Prodrug
An image of a par of scissors: the drug is essentially inactive in the dosed form and requires some chemical change in order to become pharmacologically active against its efficacy target.
The product icons (in blue) display the following information
Oral delivery
An image of a capsule.
Parenteral delivery
An image of a syringe.
Topical delivery
An image of an ointment tube.
Some drugs are dosed in multiple forms, so this is why we haven't collapsed these down to a single state. Also this icon actually represents the absorption route (so some drug that are actually deliver orally, may in fact be sublingually absorbed).
Boxed warning
An image of a black box: this is either true or false.
Availability
An image of the letters RX: the product is available as prescription.
An image of the letters OTC: the product is available over-the-counter.
An image of the letters RX with a stripe across: the product is discontinued.
Here is a little new tip/trick within the ChEMBL interface. It's possible to search by GO term - for example, if you wanted to retrieve targets (and then easily get compounds that bind to these targets) with a particular GO annotation, it's really easy to do. So, imagine you wanted targets that were GO:0008270 (which is zinc ion binding), type this in to the search box, select the "target" search button, and you get targets retrieved that have this GO term assigned. This is really cool! PS One issue is that the leading 0s in the GO term are significant
The GPCR network are running a further modelling and docking comparative study. Below is some text from the registration web page for GPCR Dock 2013, so head over there for more details.
'Based on the previous successful assessments, GPCR Dock 2008 and 2010, and the continued interest from the community on conducting a similar assessment again, we are pleased to announce that a GPCR Dock 2013 assessment will be conducted starting at midnight (Pacific standard time) February 1st and concluding at 11:59pm (PST) on March 2nd. As before, this assessment is being performed to evaluate the current status and uncover new areas of needed development in GPCR computational biology.'
Gerard and I have just had a News & Views published in Nature Methods - link to the pdf is here. This is a commentary on a paper by Lin et al. which uses metrics derived from pharmacological similarity to cluster proteins - there are some interesting differences between the same proteins clustered by sequence similarity, anyway, here's the N&V and below is the discussed paper (pdf link here)
%A G. Van Westen
%A J.P. Overington
%D 2013
%T A Ligand’s-Eye View of Protein Similarity
%J Nature Methods
%V 10
%P 116-117
%O doi:10.1038/nmeth.2339
%A H. Lin
%A M.F. Sassano
%A B.L. Roth
%A B.K. Shoichet
%T A pharmacological organization of G protein-coupled receptors
%J Nature Methods
%V 10
%P 140-146
%D 2013
%O doi:10.1038/nmeth.2324
Please see chembl_15_release_notes.txt for full details of all changes in this release, including important schema changes!
Data changes since the last release:
We have made several major changes/additions to the data in ChEMBL_15:
Incorporation of data from the USP Dictionary of USAN and International Drug Names.
Incorporation of monoclonal antibody clinical candidates and sequences.
Creation of targets for protein complexes and protein families.
Standardisation of activity data and identification of potential issues.
Annotation of predicted compound binding domains for subset of activity data.
These data sets are described in more detail in the release notes and will also be the subject of future blog posts. In addition, we have incorporated new data from the following sources:
Open TG-GATEs
TP-search transporter database
MMV Malaria Box screening data
GSK Tuberculosis screening data
GSK deposited supplementary data
DNDi Trypanosoma brucei screening data
Harvard malaria screening data
WHO-TDR malaria screening data
Database changes since the last release:
This release of ChEMBL contains major changes to the schema and data model, particularly around the representation of protein targets.
Please see the release notes, ERD and schema documentation for more details of these changes. We will also run a series of webinars over the coming weeks, describing the new schema and the changes.
Interface changes since the last release:
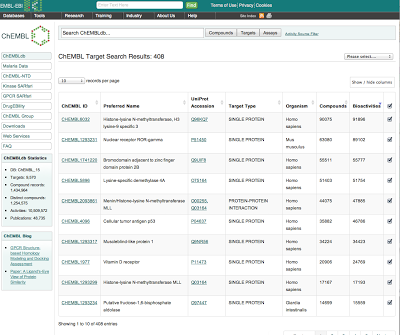
New data tables have been introduced to display search results and bioactivity data. These tables allow users to customise the display and choose which columns they want to include. By default, a standard set of columns are included in the view, but additional columns can be added by clicking on the show/hide button above the table.
A BLAST search for biotherapeutic drugs has been included on the 'Ligand Search' tab (formerly 'compound search'), allowing retrieval of protein drugs by sequence similarity.
The 'Browse Drugs' tab now includes information for monoclonal antibody clinical candidates and compounds with USANs in addition to approved drugs. Additional fields have been added and drug icons have been divided into two sets representing structure-specific information (green) and product-specific information (blue) - the latter are shown only for approved drugs.
(btw the picture above is built from ChEMBL assay descriptions - thanks to George)
I rate the InChI Software that they’ve developed, as one of the seven wonders of the modern informatics world. Long live InChI !!
One of the great things is the InChI key, a hashed form of an InChI, that is fixed length, contains only alphabetic characters (and a dash) so is metacharacter friendly, so you don’t need to worry about escaping goofy things for the web and command line text processing. There are reports of clashes for hashes, but these are very, very rare and not too difficult to deal with. For me though, one of the usability issues with InChI keys is that they do not have usable neighborhood properties, at all. By this, two compounds that only differ by one or two characters in their InChI keys are not chemically similar at all, and they shouldn’t be, it’s a hash after all.
There would be times though when it would be nice to retrieve related compounds via an InChI key search - especially in these times of google and the web. There is a simple edge case for which it’s possible to enumerate some closely structurally related InChI keys, and it turns out that this edge case is pretty common and highly useful, especially for pharmaceutically important compounds - salts.
Here is a paragraph for new people to the field, the wizards can safely skip this… Many pharmaceuticals are salts, that is they are delivered in a form where the active ingredient is ionized (either negatively if the ‘parent’ drug is an acid, or positively if the parent drug is a base). To make a compound that is overall neutral, there is a ‘salt’ component with opposite charge. These simple concepts are actually quite difficult to write, and I can think of many exceptions, but for now this will do. So acidic drugs are often salts with something like a sodium ion, and basic drugs with something with a chloride ion. An example, sildenafil is the active ingredient of the drug Revatio/Viagra - but it is dosed as the citrate salt (sildenafil citrate). The citrate has nothing to do with the bioactivity and pharmacology in vivo, but may crucial to good solubility, absorption, formulation, manufacturing, etc. So if you have sildenafil citrate as a molecule, you create an InChI key, and you would be unable then to retrieve other interesting molecules such as sildenafil itself, or other sildenafil salts, such as sildenafil malate. The usual way to deal with this in databases (like ChEMBL) is to store the data against the actual tested compound (in this case sildenafil citrate), but also to ‘salt strip’ and normalize that compound to capture the active compound sildenafil - that way you’d have two InChI keys to play with at least - one for sildenafil citrate and one for sildenafil - but you would not be able to find sildenafil malate with either of these though.
So password hashing/cracking approaches offer a simple analogous approach to this problem - and this picks up on a thread in an earlier post where we discussed rainbow tables for very large-scale databases. It turns out that if you have a plaintext dictionary of passwords, it’s really trivial to calculate the hashed forms of these, and simply do a reverse lookup from the hashed form to the plaintext form of the password. The practicality of this relies on users being human and choosing simple passwords “password1” - do a google search on “password dictionaries” and you will see how non-random and simple they are. Although the possible search space is huge, people are lazy and don’t use passwords like “dFv4%a-<
The guys and gals from the password encryption communities aren’t usually interested in chemistry, so their use of the word salt is not the same as in our chemoinformatics community - but they are sort of similar after all. The other cool thing that the password cracking community get their hands dirty with is specialist computing, and this is one area where GPU computing has made a real impact, with the performance on a specialist simple highly-scaled task (hash generation for plain-text passwords) being quite astonishing.
Plaintext -> hash function -> hashed plaintext
Plaintext.salt -> hash function -> hashed plaintext.salt
The really great thing for drugs and drug-like molecules is that although there are a huge (astronomical scale) number of potential salts for a given parent drug, the actual number used in practice is small, in the order of 50 or so (and then some of these are only used for acid drugs, some only for bases, and this property reduces the combinatorics even further). This is due to the fact that when you develop a new drug you need to explain the activity of the entire compound, not just the parent drug, and so there is a big big disincentive to developing a drug with a novel exotic salt. Also in the lab, there are a small number of salts that synthetic chemists typically make. So in the context of InChI key based searches, a useful thing to do is to enumerate all reasonable salts for a compound that is either acidic or basic, calculate the InChI keys for these, and you then have a simple synonym table that captures all reasonable representations of a drug in different salt forms. So going back to our sildenafil case - we’d have access to linking between data for sildenafil malate and sildenafil citrate which is a pretty cool thing to do.
As an interesting exercise for the reader, there are some cases that require some careful consideration - atorvastatin for instance, this is a mono-anion (carries a single negative charge) and is dosed as the calcium salt - calcium is a di-cation (i.e. It has two positive charges), so to maintain neutrality, there are two atorvastatins for one calcium (even though the USAN is atorvastatin calcium and thereby doesn’t encode the stoichiometry!). But given a suitable salt dictionary, it’s possible handle this as a simple case-branch.
The basic approach could also handle prodrugs (for cases like series of simple esters) but the semantic restrictions from this tempting hackery kick in horribly here for real world use, so do not try that at home!
It is tempting then to develop a web service, or toolkit for local high-performance use, that has a simple fast lookup of “salted” InChI key synonyms, given either a parent compound or a specific salt; or even something that computes these salted forms in real time (but for that you’d need the InChI of course, or you’d need to have that InChI key - InChI pair already in another dictionary). Another useful application of this would be to give these InChI key equivalence tables to google/bing etc…...
ChEMBL_15 will be released this week. As mentioned previously, there will be some major schema changes. For many users, the most significant of these will be:
1) Removal of protein-specific information (e.g., sequences/accessions) from the target_dictionary to a separate 'component_sequences' table. The target_dictionary now includes entries for protein complexes, protein families and other 'group' targets. These then link to their protein components via the target_components table.
2) Removal of the assay2target table. Each assay now links only to a single target (though this target may consist of multiple proteins in the case of a protein complex/family). Information previously included on the assay2target table (tid, confidence_score etc) is now on the assays table.
Please take some time to familiarise yourselves with the changes before integrating the new dataset. Further information will be provided in the release notes, and we will be running a webinar in the next few weeks to explain the changes.
For data managers of chemistry resources, the
maintenance of structure-based links to other chemistry resources can be a tedious
chore. The job is all the more burdensome knowing that your counterparts in
other chemistry based-resources are essentially duplicating your efforts, in
order to keep their links to your resource updated.
In an attempt to remove this duplication of
effort, and automate the processes involved, we have developed UniChem, and which is described in a recent publication.
Getting structure-based links out of
UniChem can be achieved either via the web-interface or the web services. For
automated updating, using the web-services is often the best choice. The
current set of web service methods has been designed to allow users several
options for how they might obtain links data. Below are detailed two
possibilities.
One such option would be to use the
following methods: First, query UniChem for all valid src_id’s using the ‘GetSrcIds’
method. Then, iterate through this list and retrieve, using the ‘GetSourceInfo‘
method,
all the details of these sources that you require (eg: the ‘base-url’ for
constructing links). Lastly, iterate through the src_id list once more, this
time retrieving all the mappings from your source to each of the other sources,
using the ‘GetMapping’ method. Combining the results of the second and third queries can provide you
with all the mappings from your compound identifiers to the URLs for the
compounds in the other sources. These data can be stored locally, and queried and
incorporated into a compound page when required. Periodic refreshes of these
local tables by repeating the above process would be required to pick up UniChem
updates.
Alternatively, you may wish to create links
more dynamically, using, for example, the ‘GetVerboseSrcCpdIdsFromInchiKey’ method. Using this method, compound web pages may be populated with all links as
the page is requested, after querying UniChem on the fly with the InChIKey.
Returned from this single query is a list of sources which contain valid
compound links. For each of the sources, a keyed list describes information
such as the ‘base-url’, etc. One of the keys (‘src-compound_id’) maps to an array
of src-compound_ids. Combining the ‘base-url’ with each of the src_compound_ids
gives the required links. See the example of this method in the link
immediately above.
 The GPCR network are running a further modelling and docking comparative study. Below is some text from the registration web page for GPCR Dock 2013, so head over there for more details.'Based on the previous successful assessments, GPCR Dock 2008 and 2010, and the continued interest from the community on conducting a similar assessment again, we are pleased to announce that a GPCR Dock 2013 assessment will be conducted starting at midnight (Pacific standard time) February 1st and concluding at 11:59pm (PST) on March 2nd. As before, this assessment is being performed to evaluate the current status and uncover new areas of needed development in GPCR computational biology.'
The GPCR network are running a further modelling and docking comparative study. Below is some text from the registration web page for GPCR Dock 2013, so head over there for more details.'Based on the previous successful assessments, GPCR Dock 2008 and 2010, and the continued interest from the community on conducting a similar assessment again, we are pleased to announce that a GPCR Dock 2013 assessment will be conducted starting at midnight (Pacific standard time) February 1st and concluding at 11:59pm (PST) on March 2nd. As before, this assessment is being performed to evaluate the current status and uncover new areas of needed development in GPCR computational biology.'
 We are pleased to announce the release of ChEMBL_15. This version of the database was prepared on 23rd January 2013 and contains:
We are pleased to announce the release of ChEMBL_15. This version of the database was prepared on 23rd January 2013 and contains: I rate the InChI Software that they’ve developed, as one of the seven wonders of the modern informatics world. Long live InChI !!
I rate the InChI Software that they’ve developed, as one of the seven wonders of the modern informatics world. Long live InChI !!





