-
Japanese Drug Approvals
 It's sometimes difficult to trace the approval process for drugs in 'foreign' countries - of course, foreign is relative, but nonetheless as a non-local it is difficult to know where to start. As an example, I tried a number of 'social media' approaches to find out about Chinese Drug non-proprietary naming - Quora, Google+ the ChEMBL-og and LinkedIn - LinkedIn was by far the best in terms of useful leads and information, often from 2nd or 3rd away links. Thanks to all that helped so far.
It's sometimes difficult to trace the approval process for drugs in 'foreign' countries - of course, foreign is relative, but nonetheless as a non-local it is difficult to know where to start. As an example, I tried a number of 'social media' approaches to find out about Chinese Drug non-proprietary naming - Quora, Google+ the ChEMBL-og and LinkedIn - LinkedIn was by far the best in terms of useful leads and information, often from 2nd or 3rd away links. Thanks to all that helped so far.
Anyway, here's a website in Japan which contains a definitive list of Japanese Drug Approvals, and which has sections in the English language to help non-Japanese readers/speakers.
It's the Pharmaceutical and Medical Devices Agency, Japan, website - http://www.pmda.go.jp/ with an English version at http://www.pmda.go.jp/english/
There are convenient, yearly approval summaries, in English - http://www.pmda.go.jp/english/service/list_s.html
Package inserts are here http://www.info.pmda.go.jp/info/iyaku_index.html (Japanese only).
As a general comment, I find the websites for the primary US and European drug approval agencies (www.fda.gov and www.emea.europa.eu) to be very complex to use, have unstable links (so things can move around and disappear), have no obvious site maps, or ways of retrieving data in a programmatic/hackamatic way - which given that these are obvious cases where opening up and ready availability of public/government data is surprising. The ability to link in to these sites is essential, and at the moment, can't be reliably done.
As an aside, there's a very good database in the UK - UK Medicines Information, which, unfortunately is only available to NHS employees, or other associated staff (presumably for licensing reasons?). However, a lot of the post-sign-in content is indexed in well known search engines - you just need to know what you are looking for ;) -
New Drug Approvals 2012 - Pt. II - Ingenol mebutate (PICATO®)

On Jan 23rd 2012 the FDA approved Ingenol mebutate gel for the topical treatment of actinic keratosis (AK). Ingenol mebutate (trade name: PICATO®, formally known as PEP-005) is a natural product derived from the euphorbia plant.
Actinic, or solar keratosis (Wikipedia; NIH; OMIM) is a pre-cancerous pigmented lesion on the skin, most commonly occurring in skin that has been frequently exposed to the sun. If left untreated, about 20% of cases transform into Squamous Cell Carcinoma.
Ingenol mebutate induces cell death in Actinic Keratosis. The precise targets responsible for the mechanism of action is not known, however, Ingenol derivatives (see e.g. CHEMBL346507) have been shown to have anticancer activities. Many of these derivatives have activity on several Protein Kinase C isoforms. Mechanistically Ingenol mebutate causes rapid lesion necrosis and also specific neutrophil-mediated, antibody-dependent cellular cytotoxicity
Ingenol mebutate (IUPAC: 2-Butenoic acid, 2-methyl-, (1aR,2S,5R,5aS,6S,8aS,9R,10aR)-1a,2,5,5a,6,9,10,10a-octahydro 5,5a-dihydroxy-4-(hydroxymethyl)-1,1,7,9-tetramethyl-11-oxo-1H-2,8a-methanocyclopenta [a]cyclopropa[e]cyclodecen-6-yl ester, (2Z) - or (1aR,2S,5R,5aS,6S,8aS,9R,10aR)-5,5a-dihydroxy-4-(hydroxymethyl)-1,1,7,9-tetramethyl-11 oxo-1a,2,5,5a,6,9,10,10a-octahydro-1H 2,8a-methanocyclopenta[a]cyclopropa[e]cyclodecen-6 yl (2Z) 2 methylbut-2-enoate). Ingenol mebutate is the mebutate ester of ingenol, and is a natural product isolated from the plant Euphorbia peplos. Ingenol is a diterpene, and is part of a diverse super-family of bioactive natural products - including phorbol esters, Reseniferatoxin and Gnidimacrin. Specifically, Ingenol mebutate is a member of the Ingenane family of natural products, and contains a Bicyclo[4.4.1]undecane ring system with in – out stereochemistry. Ingenol esters found in Euphorbiaceae, and were traditionally used in the treatment of tumors, migraines, parasites, gingivitis, and also as purgatives.
Its molecular formula is C25H34O6; molecular weight is 430.5 Da. It is a clear gel provided in two concentrations: 0.015% and 0.05%. These contain 150 mcg and 500 mcg of ingenol mebutate, respectively. It is applied to the lesion and the immediately surrounding skin. Pharmacokinetic studies have shown that absorption into the bloodstream (systemic exposure) is undetectable.
Picato® is produced and marketed by Leo Pharma.
The full prescribing information can be found here. -
Paper: Quantifying the Chemical Beauty of Drugs
There's a really interesting paper just published in Nature Chemistry - 'Quantifying the chemical beauty of drugs' from the group of Andrew Hopkins up at Dundee. Links to the paper are here, and an associated opinion piece in Nature here.
We'll add these descriptors to a future release of ChEMBL....
%T Quantifying the chemical beauty of drugs %A G.R. Bickerton %A G.V. Paolini %A J. Besnard %A S. Muresan %A A.L. Hopkins %J Nature Chemistry %V 4 %P 90–98 %D 2012 %O doi:10.1038/nchem.1243
-
Meeting: MGMS Cutting Edge Approaches to Drug Design, 26th April 2012

Registration for the Cutting Edge Approaches to Drug Design (CEADD) 2012 meeting is now open. This meeting, organised by the MGMS, is the latest in a highly successful series of conferences, which have been held in collaboration with the RSC-MMG. The meetings are aimed at those who have an interest in the use of computational chemistry in drug discovery and development. This includes medicinal chemists, as well as structural biologists and cheminformaticians. The emphasis is on interdisciplinarity in drug discovery and also on evolving tools and techniques and their application in understanding biological systems.
CEADD2012 will be held on Thursday, 26th April, 2012, at the School of Oriental and African Studies in Russell Square, London. More details, including the list of speakers and information on how to register, can be found at the conference website (http://www.mgms.org/CEADD2012/index.html).
Joining the MGMS is even easier than ever, and it's cheap; so go on, support the field of molecular modelling! -
Challenges of data integration
Here is an example of why data integration across resources is hard - and hard at multiple levels. It's presented as an example of where things can trip up the unwary..... In 1999, the structure of carbonic anhydrases II and IV complexed to the approved drug brinzolamide were published. These structures gave insight into how the drugs bind their targets, and also selectivity. The coordinate set we''re specifically interested in here is 1a42. 3-D complexes of ligands with proteins are of incredible importance in many areas, not least in docking method/scoring function development.
In the original paper the structure is drawn as....
 which is correct, and the same as found in wikipedia (Brinzolamide) and ChEMBL (CHEMBL220491). The structure in the PDB file however is something else, an isomer of brinzolamide - the IUPAC name of the ligand in the coordinate set is
which is correct, and the same as found in wikipedia (Brinzolamide) and ChEMBL (CHEMBL220491). The structure in the PDB file however is something else, an isomer of brinzolamide - the IUPAC name of the ligand in the coordinate set is(4R)-2-(2-ETHOXYETHYL)-4-(ETHYLAMINO)-3,4-DIHYDRO-2H-THIENO[3,2-E][1,2]THIAZINE-6-SULFONAMIDE 1,1-DIOXIDE
So instead of having the methoxy at the chain terminus there is an ethoxy, and the chain between the oxygen and the ring is one atom shorter - the oxygen has migrated one atom along. This is not brinzolamide, but it's called brinzolamide in the PDB entry (but the structure's wrong), and the paper (where the structure is right). The most likely explanation is that when the crystallographers built the topology file for the ligand they mistyped the atom name/element/whatever - this was a pain back then.
As a first question - what should PDB curators do here? Spot the error and fix the data, probably not, that's not the way that PDB works, but this post-loading, fixing and futzing around with the original data is common in other databases (e.g. ChEMBL). For me, this is the difference between an archive, an a curated resource.
The next level of ambiguity is where people try and extract the chemical structure from the PDB entry (for historical reasons there is no connection table in the PDB file); there's two general ways of doing this - 1) from the IUPAC name in the header, and 2) from the coordinates. Most workers have tackled the latter method, but working out the bonding from the coordinates is surprisingly hard to do completely correctly.
So what happens in this case? Well the AffinDB resource has this (AffinDB is a great resource for structure-based design and dockers).
So, two problems here, loss of stereochem off the ring (it is unambiguous in the 3D coords) and secondly the loss of the double bond for the thiophene - this has the side effect of introducing two new chiral centres into the molecule (so eight possible enantiomers, from the one defined structure used in the experiment). So the ligand, if converted to 3D, from the above structure could not recover the geometry as found in the database. Also the sulphonamide, which binds to the zinc in CAH will no longer be acidic (aryl sulphonamides are weakly acidic), and so the difference will have big differences in terms of inhibitor properties.
And what about PDBe? Well the structure there is this....

Which gets the double bond in the thiophene right, but introduces a spurious chiral centre at the sulphonamide nitrogen. This is quite a subtle case, since the nitrogen in a sulphonamide has a lot of sp3 character, and maybe one configuration is trapped in the crystal complex - but in solution, it will very rapidly invert and equilibrate - it is not a chiral centre.
In summary, this chain of events makes the data integration problem a hard one (for example if one wanted to query across ChEMBL, PDBe, AffinDB at least), and there are confounding statements on what the identity of a particular molecule is, and taking the PDB entry on face value would be confusing. So, data integration is hard! - 'Trust and Verify' is the mantra, but trust and verify names and synonyms even more.
-
Approvals and Attrition of Protein Kinase Inhibitors

Protein kinases are one of the 'hot' families in drug discovery, and last year (2011) had the highest number of approvals for protein kinase inhibitors ever - 4, these being Vandetanib (Zactima), Crizotinib (Xalkori), Ruxolitinib (Jafaki) and Vemurafenib (Zelboraf). Some of these are real breakthrough medicines, offering seriously ill patients improvement of life/extended life in terminal diseases. 2011 also saw the assignment of 12 new INNs/USANs for protein kinase inhibitors - these will form the cohort of drugs from which future approvals will come. It's interesting to track the approval ratios, a comparison of input to output (numbers of INNs/USANs to the number of approvals). Of course, there are some challenges with applying a simple approach such as this, since there is a delay between USAN/INN assignment and approval, but this will probably only be a significant issue when there is a large increase/decrease in a particular year. We also know that the 'time constant' between USAN/INN assignment and approval is about 2-4 years for approved drugs, so on average, at steady state, things will balance out.
Anyway, here are some graphs of the numbers, they should make sense without further explanation. One of the reasons for writing this was that I was at a talk recently where kinase inhibitors were treated as 'blessed' during development, that is, that they suffered lower than average attrition during development - the approval ratio is converging at about 0.2 (remember this number (one in five) is roughly the fraction of phase IIb to market, certainly not overall clinical attrition). The plotted number (blue triangles with green line) is the ratio of the cumulative USAN number to the cumulative approved number for that particular year). To my eye, the data doesn't really support this 'blessed' view - so unless the time between USAN/INN assignment and approval is systematically longer for protein kinases than other drug types (and even then under a steady assignment/approval rate scenario that doesn't matter). However, based on the large jump in 2010 USAN assignment 2013-2014 should deliver a bumper crop of new kinase drugs.

Comments as always welcome in the blog comments.
A small update - some numbers...
Highest Trial Phase Fraction with USAN IV (launched) 1.00 III 0.97 II 0.34 I 0.07
The phase 1 include quite a few Chinese NNs (still trying to get to the bottom of the naming scheme for these), also cancer is a little odd in that there are a lot of Phase I/II combined trials, and so the ratio of Phase I to Phase II is a little unusual.
-
New Drug Approvals 2012 - Pt. I - Glucarpidase (VoraxazeTM)
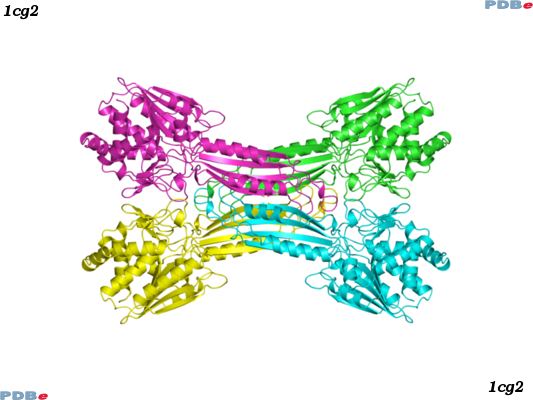
 ATC code: V03AF09Glucarpidase (Uniprot: P06621) is a carboxypeptidase produced by recombinant DNA technology in genetically modified Escherichia coli. Glucarpidase is a 390-amino acid homodimer protein with a molecular weight of 83 kDa. The crystal structure of Glucarpidase is known (PDBe: 1cg2, homotetramer form).
ATC code: V03AF09Glucarpidase (Uniprot: P06621) is a carboxypeptidase produced by recombinant DNA technology in genetically modified Escherichia coli. Glucarpidase is a 390-amino acid homodimer protein with a molecular weight of 83 kDa. The crystal structure of Glucarpidase is known (PDBe: 1cg2, homotetramer form).
The first FDA new drug approval of 2012 is Glucarpidase, approved on Jan 17th 2012. Glucarpidase (tradename: Voraxaze; formerly known as carboxypeptidase-G2 or CPG2) is a carboxypeptidase enzyme indicated for the treatment of toxic plasma methotrexate (MTX) concentrations (>1 umol/L) in patients with delayed MTX clearance due to impaired renal function.
MTX (ChEMBL: CHEMBL426) is an antifolate drug and is one of the most widely used anticancer agents. Unlike other anticancer agents, MTX can be safely administrated over a wide dose range. However, during treatment with high doses of MTX, patients may develop renal dysfunction. Since MTX is primarily cleared by renal excretion, this will lead to toxic levels of MTX. Glucarpidase acts by converting MTX to its inactive metabolites 2,4-diamino-N10-methylpteroic acid (DAMPA) and glutamic acid, providing thus an alternate route of elimination to renal excretion.

>Glucarpidase ALAQKRDNVL FQAATDEQPA VIKTLEKLVN IETGTGDAEG IAAAGNFLEA ELKNLGFTVT RSKSAGLVVG DNIVGKIKGR GGKNLLLMSH MDTVYLKGIL AKAPFRVEGD KAYGPGIADD KGGNAVILHT LKLLKEYGVR DYGTITVLFN TDEEKGSFGS RDLIQEEAKL ADYVLSFEPT SAGDEKLSLG TSGIAYVQVN ITGKASHAGA APELGVNALV EASDLVLRTM NIDDKAKNLR FNWTIAKAGN VSNIIPASAT LNADVRYARN EDFDAAMKTL EERAQQKKLP EADVKVIVTR GRPAFNAGEG GKKLVDKAVA YYKEAGGTLG VEERTGGGTD AAYAALSGKP VIESLGLPGF GYHSDKAEYV DISAIPRRLY MAARLIMDLG AGK
The recommended dosage of Glucarpidase is a single intravenous injection of 50 Units/kg.
Glucarpidase has a volume of distribution (Vd) of 3.6 L, a systemic clearance (CL) of 7.5 mL/min and an elimination half-life (t1/2) of 5.6 hours.
The full prescribing information of Voraxaze can be found here.
The license holder is BTG international Inc. -
Course - Resources for Computational Drug Discovery

A heads up on a course being held on campus here in early July - the joint EMBL-EBI and Wellcome Trust Resources for Computational Drug Discovery. The speakers are excellent, but check out the early material online for the course here. Based on previous courses, please book early to avoid disappointment ;)