-
ChEMBL Amazon Web Services

We have started to use Amazon Web Services (AWS) for a number of mini projects in the group. As part of this work we have created ChEMBL Amazon Machine Image (AMI), which we have decided to make publicly available. The ChEMBL AMI is based on the 64-bit Amazon Linux AMI, but additional comes installed with a MySQL server, which contains the ChEMBL_09 database. The benefit of making this available is that a user can have a MySQL instance up and running in a matter minutes and at almost no cost if the service is run on the AWS Free Usage Tier.
To create an instance based on the ChEMBL AMI, go through the following steps:
- Set up an AWS account (note you will need an existing Amazon account or credit card to do this).
- Login to the AWS Management Console
- Change Region to EU West (Ireland). Currently the ChEMBL AMI is only available to instances running in this region.
- Go to EC2 tab and click on Launch Instance button. The Create Instance Wizard should appear.
- On the CHOOSE AN AMI section select the Community AMIs tab and enter "chembl" in the search box. Only 1 public AMI should appear (ami-cf91a6bb), click the select button.
- On the INSTANCE DETAILS section, make sure you select Mirco instance type if you want to use the Free Usage Tier
- Proceed through CREATE KEY PAIR and CONFIGURE FIREWALL sections of the wizard and you are done
The MySQL instance will be up running and a user named chembluser (password:chembluser, we recommend you change this) has been setup with access to the chembl_09 database. So you should be able to connect to your MySQL database with the following command:
mysql -h xxxxxxxxxxxxxxxxx.compute.amazonaws.com -u chembluser -pchembluser chembl_09
(The command above assumes you have access to your instance on port 3306 from the ip range you are working from. This can be configured in the Security Groups section of the AWS Management Console).
If this proves to be a useful resource to the users of the ChEMBL data we will make publicly available AMIs for future ChEMBL database releases. -
Molecular databases and molecule complexity - part 2

Let have some examples - benzene (chembl277500) is unambiguous, it has no possibility of forming any tautomers, it cannot become protonated or lose a proton (i.e. act as a base or acid) under anything approaching physical conditions, it has no stereocenters, and furthermore has no internal degrees of freedom (it it conformationally rigid). So there is no ambiguity over calculated properties such as logP, molecular weight, etc, and you could take the structure directly from a database and do things like docking with it.
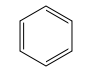
Next is pyridine (chembl266158), this has two biological forms, it is still rigid, and has no stereocenters or tautomeric forms, however, it can act as a base, and so can exist in a protonated form. These two forms have different molecular weights, overall charge and many other differences (for example, it's molecular dipole).


In particular, the binding to a receptor will be very different for these two forms, pyridine can act as a hydrogen bond acceptor, while the protonated for can't, but can act as a hydrogen bond donor - in one important factor the two forms of pyridine are opposite. The fraction of the two forms in biological systems will depend on the pH of the biological or experimental system, and also the pKa of pyridine (around 5.2 for the pKa of the conjugate acid). Typically, chemical databases will calculate and display properties for the neutral form of pyridine. Users, performing tasks such as docking, will probably need to consider both forms and dock two molecules not one.
A slightly more complicated case is 2-hydroxypyridine (chembl662), which is a classic case of tautomerism. The structure can be drawn with alternate bonding, but the two forms can rapidly interconvert. The is a small free energy difference between these two forms. The other tautomer (and the form found in solid samples of 2-hydroxypyridine) has the trivial name 2-pyridone. In solution, both forms are found, with the fractions found of each form depending on the solvent polarity.


These forms have the same molecular mass, have no stereochemical centers, but will have different calculated properties - the clogP will be different for the two forms. It is important to remember though that due to the fact they are rapidly interconvertable, they will appear to have a single logP experimentally. Again, due to the differences in hydrogen bonding potential, two forms need to be considered for a tasks like docking. What chemical databases do with cases like this varies, but typically a single tautomeric form will be used to calculate properties such as logP. What tautomer is used will depend on the particular software used.
Finally, another simple system, this time showing ambiguity over charge - 3-hydroxypyridine (chembl237847). This molecule exists in solution as an equilibrium of two physical forms, a neutral form, and a zwitterion. Calculated properties of these two forms for things like clogP will be different, the molecular weight will be the same, but again, for docking explicit consideration of the two distinct forms is required.


So hopefully, some simple examples showing that a single 2D structure of a molecule in a database can have multiple physically differing forms that can affect the calculation of properties and also have large impact on their use in modelling. Hopefully, I've also highlighted that this complexity is usually poorly handled in chemical databases (not least the current version of ChEMBL).
In the next part, we'll talk about just how complex this ambiguity is, the astronomical number of distinct structural forms possible for some molecules, and also address stereochemistry and conformational flexibility.
-
Molecular databases and molecule complexity - part 1

At one level a database of small molecules seems a really simple thing - a set of identifiers and then a 2D structure. You can then do a bunch of really cool things with this, as the large literature in the area shows. For example, one thing which is pretty common is to take a library of molecules, then 'dock' them into a protein structure, hopefully to find a novel lead; or maybe even a new use for a drug (or prediction of a side effect of a known drug). The wide availability of pipeline tools, web services connecting directly to remote databases, and so forth, makes this sort of thing really simple, and arguably too simple.
However, there are many challenges with handling normalised 2-D chemical data. One thing we have started to think about recently, is just how ambiguous a 2D representation of a structure is for typical users interested in the analysis of compound properties, docking, etc.
The problem arises from the fact that molecules are 'complex', in that a single valid 2D representation can have multiple, readily interconvertable distinct physical manefestations. These factors involving ambiguity include ionization, tautomerisation, hydration (for example, the formation of geminal hydroxy forms from aldehydes), stereoisomerism, and of course there is conformational flexibility. When a real physical experiment is done, the lowest free energy result emerges from this ensemble of possibilities.
During the registration of a molecule into a database there is a typically a series of normalisation steps that happen, in order to reduce this level of real physical world multiple structures to a simpler 'canonical' form. When one wants to use the data, a user may then need to 'enumerate' a set of possible structures in order to do anything useful with them. (of course, stereoisomers are not usually physically interconvertable. However often molecules have undefined stereochemistry when registered in a database, and for some tasks (e.g. docking) the results depend enormously on the actual stereo form, since the two enantiomers will bind to the (usually chiral) receptor with different energies, whereas other properties are identical in this case (e.g. logP).
So in summary, there is a processing step when one registers a molecule to reduce complexity in the representation, and then a processing step when one takes a molecule from a database to do something useful with it (caveat - alternatives to this general model exist).

Some molecules will have a limited (maybe even a single) number of physical forms, others will be incredibly complex and have a very large number of physical forms.
How widely appreciated is this fact - well, based on some of the questions and requests we get for ChEMBL support, I'd say not very widely, and we're thinking of ways to incorporate this into the database somehow.....
It's Easter Egg Hunt time in Cheam now.... so more tomorrow. -
April 2011 USANs
The March/April 2011 USANs have been published, these are:
USAN Research Code Structure Drug Type Drug Class Target abexinostat, abexinostat hydrochloride PCI-24781, S-78454, PCI-24781-01, S-78454-1, CRA-024781-HCl, CRA-024781-01 
synthetic small molecule therapeutic HDAC anivamersen, anivamersen sodium RB-007 
oligonucleotide therapeutic brodalumab AMG-827 monoclonal antibody therapeutic IL-17RA cabozantinib, cabozantinib s-malate XL-184, BMS-907351 
synthetic small molecule therapeutic MET RET VEGFR1 VEGFR2 VEGFR3 calaspargase pegol EZN-2285 
enzyme therapeutic - cantuzumab ravtansine huC242-DM4, IMGN-242 monoclonal antibody conjugate therapeutic mucin CanAg cenderetide CD-NP peptide therapeutic NPRB crenolanib ARO-002; CP-868,596 
synthetic small molecule therapeutic PDGFR dabrafenib, dabrafenib mesylate GSK-2118436A 
synthetic small molecule therapeutic MEK daclatasvir, daclatasvir dihydrochloride BMS-790052 
synthetic small molecule therapeutic HCV NS5A dacomitinib PF-00299804-03 
synthetic small molecule therapeutic EGFR, HER2 dasolampanel 
synthetic small molecule therapeutic delanzomib CEP-18770, CT-47098, NPH-007098 
synthetic small molecule therapeutic Proteosome dolutegravir, dolutegravir sodium GSK-1349572, S-349572, GSK-1349572A 
synthetic small molecule therapeutic HIV-1 integrase encaleret, encaleret sulfate 
synthetic small molecule therapeutic fiboflapon, fiboflapon sodium GSK-2190915A, GSK-2190915B 
synthetic small molecule therapeutic FLAP iofolastat I123 MIP-1072 
synthetic small molecule diagnostic PSMA irdabisant CEP-26401 
synthetic small molecule therapeutic H3 ixekizumab LY-2439821 monoclonal antibody therapeutic IL-17A levomilnacipran, levomilnacipran hydrochloride F-2695 
synthetic small molecule therapeutic NET SET lexibulin CYT-997 
synthetic small molecule therapeutic lumacaftor VX-809, VRT-826809 
synthetic small molecule therapeutic narnatumab IMC-RON8, IMC RON-8, RON8 monoclonal antibody therapeutic RON navarixin SCH-527123 
synthetic small molecule therapeutic CXCR1, CXCR2 olcorolimus SAR-943, SAR-943N, SAR-943-NXA, SDZ-227-943N 
natural product-derived small molecule therapeutic FKBP-12 pegadricase Uricase-PEG 20 
enzyme therapeutic - peginterferon lambda-1a PEG-IL-29, PEG-rIL-29 
protein therapeutic IL28RA pegnivacogin, pegnivacogin sodium RB-006 
oligonucleotide therapeutic rucaparib, rucaparib phosphate AG-14699, AG-014699, PF-01367338, AG-14447 
synthetic small molecule therapeutic PARP sepantronium bromide YM-155 
synthetic small molecule therapeutic BIRC5 simeprevir TMC-435; TMC-435350 
synthetic small molecule therapeutic HCV NS3/4A sirukumab CNTO-136 monoclonal antibody therapeutic IL-6 suvorexant MK-4305 
synthetic small molecule therapeutic OX1 OX2 tabalumab LY-2127399 monoclonal antibody therapeutic BAFF technetium 99m tilmanocept 1600 
synthetic small molecule diagnostic tofacitinib CP-690550, tasocitinib 
synthetic small molecule therapeutic JAK3
As I get a few moments, I'll add the missing links and structures.... Sorry the structures are goofy.
Drug TypeNumbersynthetic small molecule31natural product derived small molecule1protein1peptide1monoclonal antibody7enzyme2oligonucleotide2Cumulative total for 201145 -
ChEMBL RESTful Web Service API - Update
We are pleased to announce that we have updated the ChEMBL RESTful Web Service API (application programming interface) with some more of the features that you, the ChEMBL users requested. In particular, we have added support for the:
- Retrieval of results in JSON data format
- Searching of compounds by Standard InChiKey
- Searching of targets by UniProt and RefSeq identifiers
Sample urls:- Retrieve a compound record in JSON format - https://www.ebi.ac.uk/chemblws/compounds/CHEMBL1.json
- Search for a compound based on a given Standard InChiKey - https://www.ebi.ac.uk/chemblws/compounds/stdinchikey/QFFGVLORLPOAEC-SNVBAGLBSA-N
- Search for a target based on a given UniProt accession - https://www.ebi.ac.uk/chemblws/targets/uniprot/Q13936
- Search for a target based on a given RefSeq accession and return it in JSON format - https://www.ebi.ac.uk/chemblws/targets/refseq/NP_001128722.json
In addition to those new features we have added some example Perl and Python scripts to our documentation page. We hope that these scripts will help you in your efforts to interact with the ChEMBL API from those popular programming languages. If you have any queries or problems then please e-mail us on: chembl-help@ebi.ac.uk and we'll be happy to help.
Update: We have built some Pipeline Pilot protocols using these RESTful web services; more to come on this soon.... -
MedChemBuzz: Really good Med Chem Blog
 Just a short note to highlight a really good blog on med chem and drug discovery - MedChemBuzz - just the thing for your daily 2 hour 45 minute commute to work ;)
Just a short note to highlight a really good blog on med chem and drug discovery - MedChemBuzz - just the thing for your daily 2 hour 45 minute commute to work ;)
-
New Drug Approvals 2011 - Part XI Gabapentin enacarbil (HorizantTM)

ATC code (partial): N03AXOn April 6th, the FDA approved gabapentin enacarbil (tradename Horizant, Research Code: XP-13512, NDA 022399) for the treatment of moderate-severe forms of restless legs syndrome (RLS).
Patients suffering from RLS experience an urge to move their legs or other limbs. This urge is prompted by a painful or itchy sensation in the corresponding limb. Symptoms are most severe during phases of relaxation. Patients also sometimes have limb jerking during sleep.
Gabapentin enacarbil is a prodrug of the anticonvulsant and analgesic gabapentin (CHEMBL940). Much of the processing of gabapentin enacarbil into the active ingredient takes place in enterocytes, upon absorption in the gut. These first pass modifications are mainly mediated by non-specific carboxylesterases and via several steps of hydrolysis and yield gabapentin (the active ingredient), along with carbon dioxide, acetaldehyde and isobutyric acid. Absorption of Gabapentin into the enterocytes is likely mediated by the monocarboxylate transporter 1 (Uniprot: P53985).
The mechanism of action of gabapentin enacarbil is due entirely to gabapentin, the active component. Gabapentin binds to the a2δ subunit of voltage-gated calcium channels in vitro (Uniprot: Q9NY47). However, it is not fully known how this binding translates into a therapeutic effect. Despite its structural similarity to the neurotransmitter gamma-amino butyric acid (GABA), gabapentin has no effect on the binding, uptake or catabolism of GABA.
Gabapentin enacarbil (Smiles: CC(C(OC(OC(NCC1(CC(O)=O)CCCCC1)=O)C)=O)C, IUPAC: (1-{[({(1RS)-1-[(2-Methylpropanoyl)oxy]ethoxy}carbonyl)amino]methyl} cyclohexyl) acetic acid, InChI: 1S/C16H27NO6/c1-11(2)14(20)22-12(3)23-15(21)17-10-16(9-13(18)19)7-5-4-6-8-16/h11-12H,4-10H2,1-3H3,(H,17,21)(H,18,19), ChemSpider: 8059607) has a molecular weight of 329.39 Da. With two hydrogen bond donors and seven hydrogen bond acceptors and ACD/LogP = 2.66 gabapentin fully complies with the Lipinski rule of five. Gabapentin enacarbil is a racemate, the stereocenter is labelled with an asterisk (in the figure above).

The mean bioavailability of gabapentin enacarbil is 75% in fed state. As for conventional gabapentin, the bioavailability of gabapentin encarbil is lower if administered onto and empty stomach (42-65%). The TMAX after administration of 600mg of gabapentin enacarbil is ~7h while TMAX for conventional gabapentin is only 2h. Plasma protein binding (ppb) of gabapentin is less than 3% and the volume of distribution is 76 L. The elimination of gabapentin is mainly renal (94% recovered from urine) and renal clearance (CLr) ranges from 5-7 L/hr. While gabapentin enacarbil is extensively processed, the active ingredient gabapentin is not subject to further metabolic modifications. Compared to the parent drug Gabapentin, Gabapentin enacarbil shows an extended release and higher bioavailability. Studies showed no interactions with the major cytochrome P450 enzymes and p-glycoprotein.
The recommended dose for gabapentin enacarbil is 600mg oral, once daily.
The full prescribing information can be found here.
Gabapentin enacarbil is marketed under the name Horizant and was developed by Glaxo Smith Kline and Xenoport. -
New Drug Approvals 2011 - Pt. X Vandetanib (ZactimaTM)

ATC code: L01XE12
Vandetanib (also known as ZD-6474 and Trade name:ZactimaTM) ( IUPAC:N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine); InChI:1S/C22H24BrFN4O2/c1-28-7-5-14(6-8-28)12-30-21-11-19-16(10-20(21)29-2)22(26-13-25-19)27-18-4-3-15(23)9-17(18)24/h3-4,9-11,13-14H,5-8,12H2,1-2H3,(H,25,26,27) SMILES:COc1cc2c(Nc3ccc(Br)cc3F)ncnc2cc1OCC4CCN(C)CC4 ChEMBL:24828; ) It has the molecular formula C22H24BrFN4O2 and has a molecular weight of 475.36. It has no chiral centres. Vandetanib contains an aminoquinazoline, a very common group within a large number of protein kinase inhibitors - this mimics the adenine ring of ATP.
Vandetanib has been issued with a black box warning because it can prolong QT interval (the time between the start of the Q wave and the end of the T wave in the heart's electrical cycle. A prolonged QT interval is a biomarker for ventricular tachyarrhythmias like torsades de pointes and a risk factor for sudden death.) For this reason, Vandetanib should not be used in patients with hypocalcemia, hypokalemia, hypomagnesemia, or long QT syndrome. Vandetanib tablets for daily oral administration are available in two dosage strengths, 100 mg and 300 mg, containing 100 mg and 300 mg of vandetanib, respectively. The pharmacokinetics of vandetanib at the 300 mg dose in MTC patients are characterized by a mean clearance (Cl) of approximately 13.2 L/h, a mean volume of distribution of approximately 7450 L, and a median plasma half-life (T1/2) of 19 days.Vandetanib has a broad activity profile, showing activity against multiple tyrosine kinases including RET (Uniprot: P07949; canSAR Target Synopsis) , EGFR (Uniprot: P00533; canSAR Target Synopsis), FGFR1 (Uniprot: P11362; canSAR Target Synopsis), FGFR2 (Uniprot: P21802; canSAR Target Synopsis), FGFR3 (Uniprot: P22607; canSAR Target Synopsis), and many others, all of which are members of the Protein Tyrosine Kinase family (PFAM:Pkinase_Tyr (PF07714)). RET mutations associated with medullary thyroid cancer include C634R germline mutation in exon 11 and an additional somatic mutation (at chromosomal position 164761.0012), but the efficacy of Vandetanib is independent of the mutation status of RET. A complex structure of Vandetanib bound to RET is available (PDB code: 2ivu @PDBe)
Vandetanib tablets for daily oral administration are available in two dosage strengths, 100 mg and 300 mg, containing 100 mg and 300 mg of vandetanib, respectively. The pharmacokinetics of vandetanib at the 300 mg dose in MTC patients are characterized by a mean clearance (Cl) of approximately 13.2 L/h, a mean volume of distribution of approximately 7450 L, and a median plasma half-life (T1/2) of 19 days.Vandetanib has a broad activity profile, showing activity against multiple tyrosine kinases including RET (Uniprot: P07949; canSAR Target Synopsis) , EGFR (Uniprot: P00533; canSAR Target Synopsis), FGFR1 (Uniprot: P11362; canSAR Target Synopsis), FGFR2 (Uniprot: P21802; canSAR Target Synopsis), FGFR3 (Uniprot: P22607; canSAR Target Synopsis), and many others, all of which are members of the Protein Tyrosine Kinase family (PFAM:Pkinase_Tyr (PF07714)). RET mutations associated with medullary thyroid cancer include C634R germline mutation in exon 11 and an additional somatic mutation (at chromosomal position 164761.0012), but the efficacy of Vandetanib is independent of the mutation status of RET. A complex structure of Vandetanib bound to RET is available (PDB code: 2ivu @PDBe)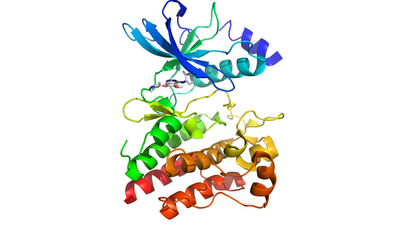 The prescribing information can be found here. Vandetanib is a product of AstraZeneca
The prescribing information can be found here. Vandetanib is a product of AstraZeneca









{kind=link}
{kind=link}