An interesting compound synonym error recently caught our eye. It is in the existence of a common corrupted synonym in the research code for Imatinib - the correct research code is STI571, but it is surprisingly commonly found as ST1571 where the capital I has been replaced by the number 1. In many fonts of course, these are identical, or very close, in rendering (as is the lower case l). You may think that it is a rare error, but I guess once made, it has widely propagated across the web. Of course ST1571 could be anything, a hammer, a breed of carrot, etc, but most of the references to ST1571 are in fact to Imatinib.
As of 16th November, the google search counts are 84,600 for STI571, and 18,100 for ST1571. And now, damn, I've further propagated this confusion of the co-occurrence relationship with these words and Imatinib!
So, this is mere bagatelle, a minor frippery, a rabbit-hole of nihilism? - but these things are important when you want to reliably aggregate and mine across free format Interweb text.....
FDA approved drugs have now been added to the compounds table*. Some drugs (e.g., biotherapeutics) do not have a structure/molfile, and not all drugs have bioactivity data associated with them. Further information for these drugs (e.g., mechanism of action) will be added in subsequent releases.
Parent compounds have been generated by removing the salt component from any compounds tested as a salt form. Both the parents and the salt forms are recorded in the compounds table and a new table: molecule_hierarchy shows the relationship between them.
ChEMBL identifiers (chembl_id) have been added to the compounds, target_dictionary, assays and docs tables. These take the form 'CHEMBL' followed immediately by an integer (e.g., CHEMBL941) and are used on the interface. Small molecules within the database will still have a ChEBI ID, and protein targets a UniProt accession, in addition.
The interface now uses chembl_id for compounds, assays and targets. Old URLs (e.g., using chebi_id/assay_id/tid) will continue to work, however we recommend using the chembl_id when linking to the ChEMBL interface.
The ChEMBL Team *The identification and loading of the FDA approved compounds in the ChEMBL database is part of a larger process of integrating drug and clinical candidate information into the ChEMBL database. This process has not not been completed, so please expect enhancements to the underlying schema and interface in future releases of the ChEMBL database.
Unfortunately, I cannot go to the SMR meeting - I will be ill (probably), in a hotel (hopefully), maybe on the beach (certainly) at the Zing Structural Biology Conference. The graph above answered an important question for me.
Ceftaroline Fosamil is a semisynthetic antibacterial of the cephalosporin class of beta-lactams, which are originally identified in 1948 from the Cephalosporum/Acremonium. Ceftaroline Fosamil is the phosphamide prodrug of the bioactive Ceftaroline. Like other drugs in the same class, the bactericidal action of Ceftaroline is mediated through covalent binding to essential penicillin-binding proteins (PBPs) in the bacteria wall. In particular, ceftaroline is bactericidal against S. aureus, including methicillin-resistant S. aureus (MRSA), due to its affinity for PBP2a (Uniprot: Q53707, ChEMBL: 19669), the type of PBP produced by MRSA and not well inhibited by other antibiotics such as methicillin (ChEMBL: 116716), oxacillin (ChEMBL: 156432), penicillin, and amoxicillin (ChEMBL: 657723). Ceftaroline is also active against S. pneumoniae due to its affinity for PBP2x (Uniprot: P14677, ChEMBL: 102467).
Ceftaroline Fosamil is a large 'small-molecule' semisynthetic prodrug (Molecular Weight of 685.7 g.mol-1 for Ceftaroline Fosamil itself and 762.7 g.mol-1 for the monoacetate salt), slightly lipophilic and soluble in water. Following injection, Ceftaroline Fosamil has a volume of distribution of 20.3L, a low plasma protein binding (ppb) of 20%, an elimination half-life of 1.6hr and a plasma clearance of 9.58 L/hr. Ceftaroline Fosamil is primarily eliminated by the kidneys (88% of the dose is recovered in urine) and mainly as the active metabolite ceftaroline (64% as ceftaroline and 2% as an inactive metabolite). Ceftaroline is not an inhibitor or substrate of the major cytochrome P450 isoenzymes. The recommended dosage of Ceftaroline Fosamil is 600mg every 12 hours by intravenous infusion administrated over an hour.
The full prescribing information can be found here. Like other cephalosporins, Ceftaroline Fosamil structure (6R,7R)-7-{(2Z)-2-(ethoxyimino)-2-[5-(phosphonoamino)-1,2,4thiadiazol-3-yl]acetamido}-3-{[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl}-8-oxo-5-thia-1azabicyclo[4.2.0]oct-2-ene-2-carboxylate contains a cyclic amide (the beta-lactam ring) fused with a six member ring (the cephem ring). Another notable feature of Ceftaroline Fosamil is the thiazolylthio group, which is thought to be crucial for the activity against MRSA.
NAME="Ceftaroline Fosamil"
TRADEMARK_NAME="Teflaro"
ATC_code= NA
SMILES="CCO\N=C(/C(=O)N[C@H]1[C@H]2SCC(=C(N2C1=O)C(=O)O)Sc3nc(cs3)c4cc[n+](C)cc4)\c5nsc(NP(=O)(O)O)n5"
InChI="InChI=1S/C22H21N8O8PS4/c1-3-38-26-13(16-25-21(43-28-16)27-39(35,36)37)17(31)24-14-18(32)30-15(20(33)34)12(9-40-19(14)30)42-22-23-11(8-41-22)10-4-6-29(2)7-5-10/h4-8,14,19H,3,9H2,1-2H3,(H4-,24,25,27,28,31,33,34,35,36,37)/p+1/b26-13-/t14-,19-/m1/s1"
ChemDraw=Ceftaroline_Fosamil.cdx
Registration for the 2011 residential ChEMBL Training course, running from the 14th to the 18th of Feburary, is now underway, further details can be found at this link.
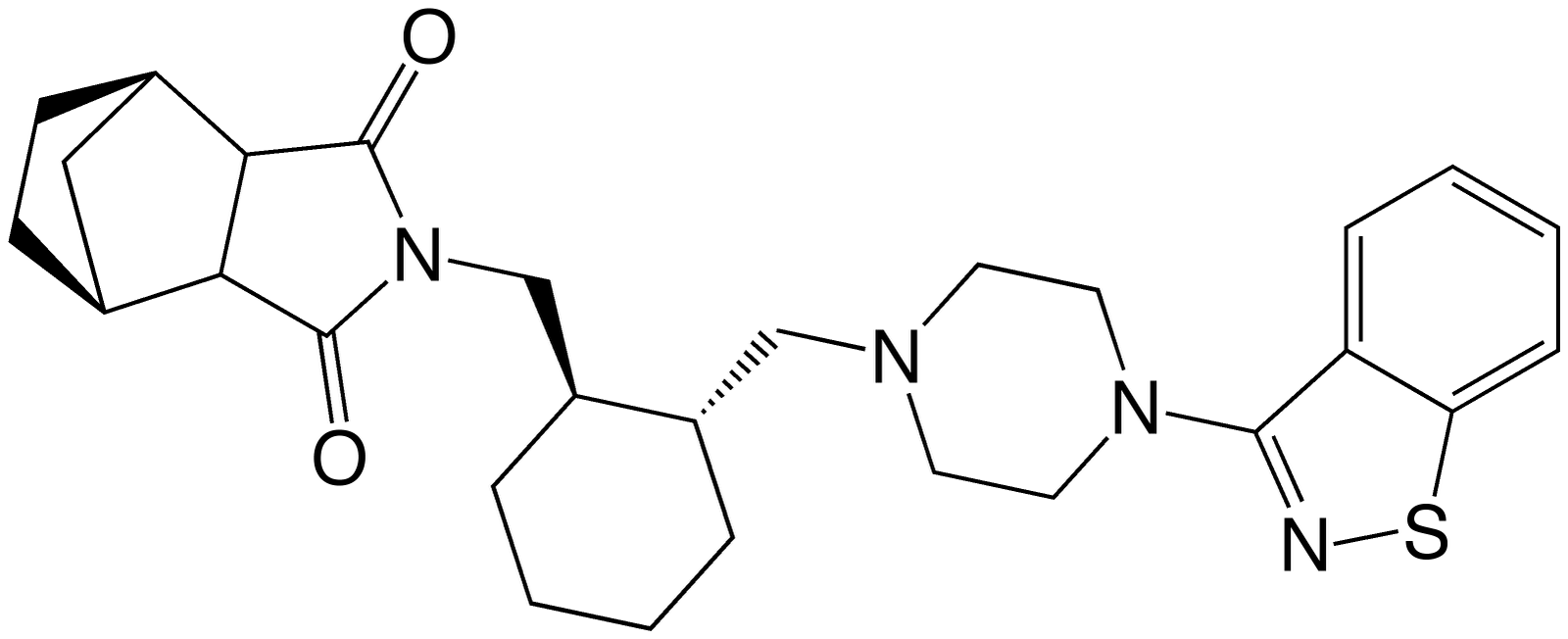
On October 28th 2010, the FDA approved Lurasidone (Tradename:Latuda) (Lurasidone is also known by the research code SM-13,496). Lurasidone is an atypical antipsychotic agent indicated for the treatment schizophrenia.
Lurasidone displays broad polypharmacology against a wide range of rhodopsin-like aminergic GPCRs, acting as an antagonist with high affinity at dopamine D2 receptors (Uniprot: P14416, ChEMBL: 72) (Ki of 1 nM), serotonin 5-HT2A (Uniprot: P28223, ChEMBL: 107) (Ki of 0.47 nM) and 5-HT7 receptors (Uniprot: P34969, ChEMBL: 10209) (Ki of 0.49 nM), and with moderate affinity at alpha-2C adrenergic receptors (Uniprot: P18825, ChEMBL: 218) (Ki of 10.8 nM) and at alpha-2A adrenergic receptors (Uniprot: P08913, ChEMBL: 52) (Ki of 40.7 nM). Lurasidone acts also as a partial agonist at serotonin 5-HT1A receptors (Uniprot: P08908, ChEMBL: 51) (Ki of 6.4 nM) and exhibits little or no affinity for histamine H1 (Uniprot: P35367, ChEMBL: 127) and muscarinic M1 receptors (Uniprot: P11229, ChEMBL: 61) (IC50 > 1000 nM and IC50 > 1000 nM, respectively). The efficacy of Luasidone is thought to be primarily related to the D2 and 5HT2A antagonism. All atypical antipsychotics display this complex polypharmacology.
Lurasidone is a synthetic small-molecule drug (Molecular Weight of 492.7 g/mol for Lurasidone itself and 529.14 g.mol-1 for the dosed HCl salt), is fully Rule-of-Five compliant, lipophilic and very slightly soluble in water.
Lurasidone has low systemic bioavailability (9-19%), and a high volume of distribution of 6173L, and displays high plasma protein binding (ppb) of ~99%. The half life is 18 hours, and steady-state plasma levels are reached 7 days after starting regular dosing. Lurasidone is predominantly metabolized by CYP3A4 into four major metabolites (two active metabolites and two 'inactive') - metabolites include hydroxylation of the nornbornane ring, N-dealkylation and S-oxidation. The apparent clearance is 3902mL/min, with the bulk of the drug being excreted in the feces. Dosage is oral, with a recommended starting dosage is 40 mg once daily (equivalent to 81umol), with a recommended maximum dosage of 80 mg daily.
Lurasidone is a chiral benzoisothiazol derivative - the benzoisothiazol is the fused five-six dual ring structure on the right of the figure above. Its structure (3aR,4S,7R,7aS)-2-{(1R,2R)-2-[4-(1,2-benzisothiazol-3-yl)piperazin-1-ylmethyl] cyclohexylmethyl}hexahydro-4,7-methano-2H-isoindole-1,3-dione contains an imide heterocyclic and a piperazine functional group. The central piperazine nitrogen is basic. The chemical structure, properties and pharmacology are similar to Ziprasidone (Trademark:Geodon).
NAME="Lurasidone"
TRADEMARK_NAME="Latuda"
ATC_code= NA
SMILES="O=C1C2[C@@H]3CC[C@@H](C3)C2C(=O)N1C[C@@H]4CCCC[C@H]4CN5CCN(CC5)c6nsc7ccccc67"
InChI="InChI=1S/C28H36N4O2S/c33-27-24-18-9-10-19(15-18)25(24)28(34)32(27)17-21-6-2-1-5-20(21)16-30-11-13-31(14-12-30)26-22-7-3-4-8-23(22)35-29-26/h3-4,7-8,18-21,24-25H,1-2,5-6,9-17H2/t18-,19+,20-,21-,24?,25?/m0/s1"
ChemDraw=Lurasidone.cdx
The full prescribing information can be found here.
Lurasidone has a boxed warning (colloquially known as a 'black box').




 The next Society for Medicines Research meeting is on the 9th December 2010 at the National Heart and Lung Institute, Kensington, London. These are my favourite day meetings, cheap, well organised and very applied to actual drug discovery. This meeting there are some great talks.
The next Society for Medicines Research meeting is on the 9th December 2010 at the National Heart and Lung Institute, Kensington, London. These are my favourite day meetings, cheap, well organised and very applied to actual drug discovery. This meeting there are some great talks.
 Like other cephalosporins, Ceftaroline Fosamil structure (6R,7R)-7-{(2Z)-2-(ethoxyimino)-2-[5-(phosphonoamino)-1,2,4thiadiazol-3-yl]acetamido}-3-{[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl}-8-oxo-5-thia-1azabicyclo[4.2.0]oct-2-ene-2-carboxylate contains a cyclic amide (the beta-lactam ring) fused with a six member ring (the cephem ring). Another notable feature of Ceftaroline Fosamil is the thiazolylthio group, which is thought to be crucial for the activity against MRSA.
Like other cephalosporins, Ceftaroline Fosamil structure (6R,7R)-7-{(2Z)-2-(ethoxyimino)-2-[5-(phosphonoamino)-1,2,4thiadiazol-3-yl]acetamido}-3-{[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl}-8-oxo-5-thia-1azabicyclo[4.2.0]oct-2-ene-2-carboxylate contains a cyclic amide (the beta-lactam ring) fused with a six member ring (the cephem ring). Another notable feature of Ceftaroline Fosamil is the thiazolylthio group, which is thought to be crucial for the activity against MRSA. 



 The November batch of USANs have recently been announced.
The November batch of USANs have recently been announced.