Today is election day in the UK, and so it's ones duty to vote, and then spend the rest of the day thinking about just how cool science is.
I was thinking the other day on the train, that we don't always want drugs that are benign, of course we always want drugs that are safe and predictable in action; but mechanistically we sometimes want to do harm, kill a cell, wipe it out entirely - let's call these malign. Of course, this socially responsible expression of this desire to obliterate life with drugs is in the development of antibiotics (bacteria, viruses, fungi and parasites) and also anti-cancer agents. The other system we sometimes want to modulate to cause specific harm is via activation of the immune system - priming it to attack non-self (pathogen) cells, or that limbo-land of pseudo-self cancer cells.
Anyway, this thinking of drugs as either (simplistically) malign or benign was triggered by a mail from a new collaborator interested in antiparasite agents identified via drug repositioning. How could you come up with a prioritised and annotated set of current drugs (and their associated cognate targets) that are biased to killing certain cells?
There are many approaches to this problem, here is just one... Given that the majority of drugs try to do no harm, and restore normal function to the recipient, screening most 'benign' drugs would be expected a priori to do little. Do we really want to find a way to balance sugar metabolism in a plasmodium and cure it's diabetes? - of course not, so screening known antidiabetic agents is less likely to reveal a cidal phenotype.
sporadic binding of a non-cidal drug/target to a new cidal target in the pathogen.
a benign-malign target switch occurs - so a context (organism) dependent switch happens - a nice example here is the statins, which were first isolated and characterised as anti fungal natural products, and now of course are used as cholesterol lowering agents in humans. It’s left as an exercise for the reader to answer why statins aren’t used as antifungals.
There are exceptions to all such half-assed approximations. The two most obvious are
So, let’s have a high level go at classifying drugs as malign or benign in intent. I’m on a train, so it will need to be a quick and dirty approach…. Use the WHO ATC classification! Classes J, L and P ('Antiinfectives for systemic use', 'antineoplastic and immunomodulating agents', and 'anti-parasitic products, insecticides and repellents' respectively) are clearly enriched in malign drugs (and malign targets), and certain subclasses of other top level classes are also malign - e.g. D01, D06 and G01 for example.
So, we can rapidly (and approximately) classify drugs into malign and benign classes using the ATC, and via simple extension produce sets of malign and benign targets. We can even address some of the more-likely benign to malign switches, for example using sequence similarity to identify similar architectures/mechanisms. As an example, quite a few antithelmintics work by binding to ion channels and causing paralysis, finding ATC classes of ion-channel blockers is straightforward.
As promised, we would like to provide the answer to the Easter egg hunt competition and announce the winner. Exactly seven hours after publishing the blog post we received a comment with the correct answer. The author of the comment was Matt Swain, who runs his blog about cheminformatics.
You can verify the correctness of his answer by visiting the password protected link in the original post. The password is:
We are pleased to announce a new round of resource-specific webinars that will be given in May and June 2015. These four webinars will cover UniChem, ChEMBL, MyChEMBL (the ChEMBL Virtual Machine) and the ChEMBL Web Services.
ChEMBL Walkthrough, 4pm BST, 20th May 2015, Register Here
MyChEMBL Walkthrough, 4pm BST, 10th June 2015, Register Here
ChEMBL Web Services, 4pm BST, 17th June 2015, Register Here
For those of you who can't make these days/times, each 1 hour long webinar will be video recorded and will be available to watch on YouTube afterwards. Additionally, we will make the slides available for download.
The video for last month's SureChEMBL webinar can be found here (and part 2 here).
For more information about the webinars, or to suggest other topics to cover, please contact chembl-help@ebi.ac.uk.
Easter is coming and for all those, who don't know what to do with their spare time and fancy entering a little competition, we've prepared a small challenge.
Easter Egg?
In software development, an Easter egg is funny (but harmless) and undocumented feature hidden from users in unusual places. Excel 97 has its Flight Simulator, FireFox about:robots address and Debian's apt-get has a moo command. The ChEMBL web services has now joined this list and we invite you to find its hidden feature and share with others.
But why?
We would like to encourage you to look at the source code of our web services. Reading code is essential developer skill, as it helps in understanding how the code works. This can lead to the development of new software and/or improve an existing codebase. After skimming through the code, hopefully you will agree that it is well written and easy to extend. Let us know if you disagree, either by emailing us or creating a GitHub issue. We promise, there are no dragons there, only an Easter egg, which should be easy to find after reading the code.
How?
The first person to comment on this post with a URL revealing the Easter egg (web service URL, hmm hint maybe?), will be honoured with a mention in a future blog post. As a 'guarantee', that the Easter egg exists now, we provide the following password protected URL - we will reveal the password, the winner and more details in the follow up blog post. Do you think you can do it? Check it out!
Is this an April fool joke?
No. The joke is in having an Easter egg, not in finding it.
We have a long-standing interest in finding clinical stage compounds from the literature - and it turns out that the peer reviewed literature is pretty useless, by the time something is published and appears in print it is old news, and although reviews of particular areas or targets are useful in capturing a snapshot, they are not really useful in decision support - using data to inform future experiments and investment. So these things need to be databased and online to be of much use.
So, we have a pretty big set of clinical stage kinase inhibitors that we've gathered from a wide variety of sources - this is the subject of a paper we're currently writing up, so I won't bore you with how we got the data, well, not right now.
We've posted a couple of times before about the transition of names, or classes of names as compounds go through development and approval - a search of the ChEMBL-og will show you these. But here's something hot off the press.
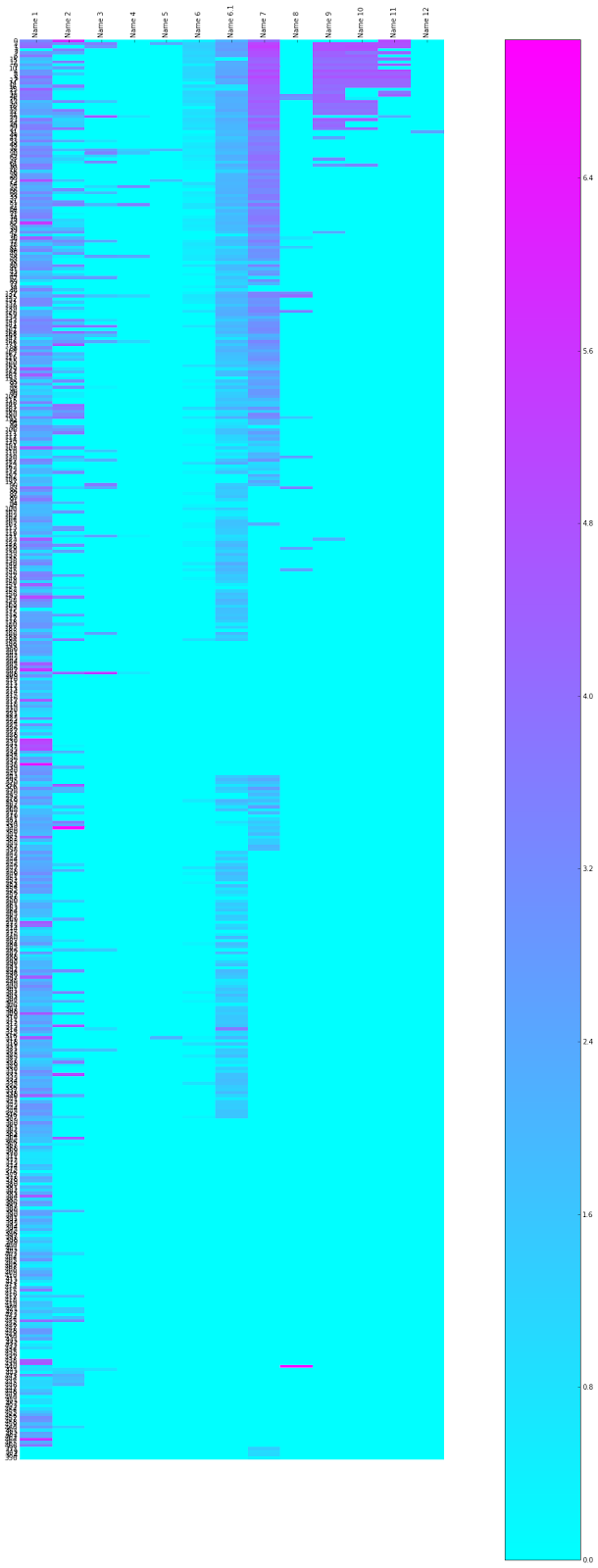
Each row is a distinct kinase inhibitor, each column is a synonym or identifier for that compound. Columns 1 to 5 are research code type names (e.g. UK-92480), with the first column being the one we use as the primary identifier, Column 6 is the InChI key, Column 6.1 is the CAS Registry number, 7 is the USAN/INN 8 is a deprecated USAN/INN or another common trivial name, Columns 9 to 12 are trade names (in case you are wondering, row 12 is of the Chinese tradename of a compound). The cells are coloured by the log(10) of the count of the number of times the name occurs in a google search - pink high, blue low (or there is no synonym of that class for that compounds). There are some false positives, where the name is unusually common, so it matches the name of something unrelated to a kinase inhibitor or therapeutic application.
It's interesting to see the diversity of names increasing as a compound becomes a launched drug, but also the broad coverage for many of these compounds with hits to the InChI key - and also for what fraction compound structures are known.
As many of you know, SureChEMBL taps into the wealth of knowledge hidden in the patent documents. More specifically, SureChEMBL extracts and indexes chemistry from the full-text patent corpus (EPO, WIPO and USPTO; JPO titles and abstracts only) by means of automated text- and image-mining, on a daily basis. We have recently hosted a webinar about it which turned out to be very popular - for those who missed it, the video and slides are here.
Besides the interface, SureChEMBL compound data can be accessed in various ways, such as UniChem and PubChem. The full compound dump is also available as a flat file download from our ftp server.
Since the release of the SureChEMBL interface last September, we have received numerous requests for a way to access compound and patent data in a batch way. Typical use-cases would include retrieving all compounds for a list of patent IDs, or vice versa, retrieving all patents where one or more compounds have been extracted from. As a result, we have now produced this so-called map file which connects SureChEMBL compounds and patents.
There is a total of 216,892,266 rows in the map, indicating a compound extracted from a specific section of a specific patent document. The format of the file is quite simple: it contains compound information (SCHEMBL ID, SMILES, InChI Key, corpus frequency), patent information (patent ID and publication data), and finally location information, such as the field ID and frequency. The field ID indicates the specific section in the patent where the compound was extracted from (1:Description, 2:Claims, 3:Abstract, 4:Title, 5:Image, 6:MOL attachment). The frequency is the number of times the compound was found in a given section of a given patent. More information on the format of the file in the README file.
How many compounds and patents are there?
There are 187,958,584 unique patent-compound pairs, involving 14,076,090 unique compound IDs extracted from 3,585,233 EP, JP, WO and US patent documents - an average of ~52 compounds per patent. The patent coverage is from 1960 to 31-12-2014 inclusive. Here's a breakdown of the patents in the map per year and patent authority:
Are these all the compounds and patents in SureChEMBL?
Technically, no - in practice, yes. We excluded chemically annotated patents that are not immediately relevant to life sciences, such as this one. For the filtering, we used a list of relevant IPCR and related patent classification codes. At the same time, we excluded too small, too large, too trivial compounds, along with non-organic and radical/fragment compounds.
Are these compounds genuinely claimed as novel in their respective patents?
Automated methods to assess which are the important and relevant compounds in a pharmaceutical patent is a field of research and one of our future plans. For now, the map file include all extracted chemistry mentioned in all sections of a patent, subject to the filters listed in the previous section. A quick and effective trick to filter out trivial and/or uninformative compounds is to use the corpus frequency column and exclude everything with a value more than, say, 1000. Note that, in this way, you will also exclude drug compounds such as sildenafil, which are casually mentioned in a lot of patents. You could also look for compounds mentioned only in claims, description or images sections by filtering by the corresponding field ID.
What can I do with this?
Well, you can start by 'grepping' for one or more patent IDs or SCHEMBL IDs or InChI keys, followed by further filtering. Many of you will choose to normalise the flat file into 3 database tables (say compounds, documents and doc_to_compound) for centralised access and easy querying.
For example, to find the patents the drug palbociclib has been extracted from:
Any plans to update this map file?
New patents and chemistry arrive and are stored to SureChEMBL every day. We are planning to release new versions and incremental updates of the map file every quarter, in sync with the update of the compound dump files.
I couldn’t find my compound / patent - this compound should not be there
Don’t forget this an automated, live, high-throughput text-mining effort against an inherently noisy corpus such as patents. We are constantly working on improving data quality. If you find anything strange, let us know.
Can I join more metadata, such as patent assignee and title?
Obviously your first port of call would be the SureChEMBL website for patent metadata, but other services you may wish to use include the EPO web services for programmatic access. Is there anything else? Errr, yes. Watch this space for another post on storing and accessing live SureChEMBL data, behind your firewall. The SureChEMBL Team
We have mentioned Beaker (a.k.a the ChEMBL cheminformatics utility web service), several times on the blog (here, here and here), but have not devoted an entire post to Beaker. Well, here it is.
Beaker - what's this?
It's a small utility, that makes chemistry software available securely over https. You no longer need to install a chemical toolkit in order to convert your molfile to SMILES or calculate descriptors. If you have an internet connection (if you can read this, chances are you do), you can use Beaker. We recommend you head over to the interactive online documentation (https://www.ebi.ac.uk/chembl/api/utils/docs), to see the full list of functionality it offers and try it with your own data.
Which toolkits are used by Beaker?
Under-the-hood Beaker is exposing the functionality of the RDKit cheminformatics library. Beaker's optical structure recognition methods use the OSRA library.
Do I need an API Key?
As long as you are making no more than 1 request per second, you do not need an API key. Beaker provides standard set of response headers to inform about rate limiting:
There is also one custom header:
This lets you know how you have been authenticated. The default authentication is IP-based, which means that if any other person uses Beaker from the same IP, it will affect your rate limit. This is why having your own API key can be useful - no one can 'steal' your rate limit and it will be slightly higher than default as well. If you need a key, just write to us.
I tried it and it doesn't work...
Before contacting us and submitting bug report, please try a few things first:
1. Submit data viahttps://www.ebi.ac.uk/chembl/api/utils/docs. It it works there, see point 2.
2. Check data encoding. Unlike the ChEMBL data web services (where you should use percent encoding as described in the previous blog post), if you are accessing Beaker viaGET, then all data provided should be base64 encoded. This is why, if you want to use GET to convert 'CCC' SMILES to molfile this link won't work: https://www.ebi.ac.uk/chembl/api/utils/smiles2ctab/CCC.
'CCC' has to be base64 encoded firstand base64('CCC') == 'Q0NDQw==',
so the valid link is https://www.ebi.ac.uk/chembl/api/utils/smiles2ctab/Q0NDQw==. Our online documentation will do encoding for you, and present what URL was really executed:
3. Use POST where possible. GET requests are nice, because everything gets included into URL, so you can embed such a URL in a blogpost, like we just did. One issue with GET is that there is often a maximum number of characters you can send, although this does depend on server setup. If you would like to use Beaker from ChEMBL servers for example, your link can't exceed 4000 characters. Base64 encoding will make any parameter about 1/3 longer. So for example, if you would like to send an image, in order to perform Optical Structure Recognition (OSRA), it's very hard to find a valid, good quality image, that is less than 1.2 Kb in size, so in that case using GET is not a good idea. Also, do not forget you can use curl to submit your POST requests. Below we provide some examples of how to access Beaker via POST with curl:
4. If using GET, check what type of base64 are you using. Standard implementation of base64 use the following characters:
[a-zA-Z0-9+/]
Those two last signs ('+' and '/'), are not url-safe as they have special meaning in URLs. This is why Beaker uses url-safe version which substitutes '-' instead of '+' and '_' instead of '/' in the standard base64 alphabet. For reference, please click here.
Does ChEMBL python client library work with Beaker?
Yes, and even more it adds enough syntactic sugar to make it feel like your are using locally installed chemical toolkit. For example, look how easy it is, to compute maximum common substructure from three compounds, given as SMILES strings:
You can install the python client library by using 'pip install chembl_webresource_client' or download it here and expect more examples in a future blog post.
Does it work without the client?
Of course it does. You have already seen an example of how to use Beaker from JavaScript (using the online documentation) and python (using the client library). But because curl is very common tool, available on many platforms, you can execute calls to Beaker from your command line in bash. Bash has a very cool feature called pipes, so you can chain the output of one command to the input of another. This way you can mix calls to our data web services with Beaker calls. As an example let's assume that you have a photo of a compound. This could be a scan of the paper document, such as patent or a photo of a conference poster taken using your mobile phone, but it has to have decent resolution and quality:
If we would like to find the compound in ChEMBL that is most similar to the one recognized from the image above, we could use this line of bash script:
The script may look a bit hackish, but this is because we wanted to only use standard command line tools, that can be found on OSX and Linux systems. In production, we would never use grep, sed and awk to parse JSON because this is bad (instead we encourage you to try jq), but we wanted to show a nice example of using pipes to combine different tools. Anyway, the end result of running this command will be open the following page in your browser: https://www.ebi.ac.uk/chembl/api/data/image/CHEMBL2107150.
Is Beaker open source software, can I see the code?
This also means that you can deploy your own local Beaker version. Reasons why you might like to do this include:
1. You don't want to rely on availability of ChEMBL web services or care about rate limiting.
2. You don't want to send proprietary compounds to a public service.
3. You would prefer to install your own chemical toolkit (on only 1 machine), and access its servicesover http(s). How do I cite Beaker?
When reviewing our old web services documentation (https://www.ebi.ac.uk/chemblws/docs), you will notice some methods can be accessed by both: GET and POST. One thing these methods have in common is that they all accept SMILES as search parameter. Why? Well, it turns out there are some SMILES, that can not be handled via GET, when using old web services. Take this SMILES as an example: [Na+].CO[C@@H](CCC#C\C=C/CCCC(C)CCCCC=C)C(=O)[O-] and you will see, that you can't construct a valid URL using 'https://www.ebi.ac.uk/chemblws/compounds/smiles/' as a base. To 'GET' around this issue, you will need to use POST. This is a bit sad, as it means you can not put a link to such a compound on your blog or ask about it on chemistry forum or send it to your friend via Skype :( What's more, if you would like to use POST for a non-SMILES method (e.g. get all assays by ChEMBL ID), you would also be out of luck.
When reviewing the documentation of the new web services (as we asked you in the previous blog post), you will probably notice, that none of the methods mention POST support. What does it mean? First of all, it means that you can achieve everything using GET. You can easily retrieve data for the SMILES string above using GET, you just have to be careful. Placing a SMILES strings in URLs can be tricky. This is because they often contain characters that are not allowed in URLs and should be encoded, according to the URI standard. Encoded how? The standard mechanism to encode URLs is called percent-encoding and it is widely accepted by modern web browsers and other web tools, such as curl or wget.
In fact, some browsers hide the percent-encoding from users, as we will see soon. So the percent-encoded URL of our molecule looks like this:
Much better, only one sign, #, is encoded! What if you try to open the second link in browser? In Firefox this should work. All other popular browsers will return 404 - not found and curl will complain:
So what to do?
1. These are characters, that always have to be encoded:
% change to %25, because percent is a special 'escape' character in percent encoding.
# change to %23, because not encoded hash sign is an indicator of the Fragment identifier part of URL, which is only used in browser and is not send to server.
\ change to %5C, because all browsers apart from Firefox are changing not encoded backslash to forward slash as explained here.
2. This is a character, that can not be encoded:
Forward slash / can not be encoded as %2F because our Apache server configuration will return a 404 for URLs containing %2F. The security reasons are described in the Apache documentation. This is particularlyannoying, as it means that you can not use online percent-encoders if your SMILES contain forward slashes.
How to make your life with SMILES simpler?
1. Use Firefox - this browser does a great job when it comes to encoding. Although FF is unable to guess in which context you are using % and #, so you will still encode those two characters before pasting it into the address bar, but the rest of special characters will be encoded correctly, so then you can copy the URL from Firefox to other browsers and it will just work. 2. Use cURL - just as with Firefox, you have to encode % and #, but the rest will be handled properly, you just have to use '-g' flag, which switches off the URL globbing parser:
This is huge! But, more importantly, the SMILES looks like it's already url-encoded, as it contains %23 and %29, which will be interpreted as reserved characters and translated to # and ), when pasted into a browser address bar. To prevent this from happening, we must first percent-encode the percent sign, to ensure they are interpreted literally. Percent is encoded as %25 and after conversion our URL will look like this (this URL contain many backslashes so it will only work in Firefox):
It should be noted, that the URL is 1666 characters long, which is below the maximum allowed URL length in our Apache setup (4000 if your were wondering). Even if we encode all the special characters, we will end up with the following URL (this time valid in all modern browsers):
which is 2478 characters long and still lower than 4000 limit. This means we can use GET to retrieve every ChEMBL compound based on its SMILES string.
What about POST?
OK, so SMILES can be passed via GET, can POST still be used? Yes! For every single method you can use POST as well. This is important, as it is possible to create very long URLs, by chaining together multiple filters and other parameters, such us pagination and ordering. This is why all new web services methods (even those, which don't expect any SMILES), can be executed using POST. In fact, our updated Python client (blog post coming soon!), is using POST for almost everything. Just keep in mind, that in order to use POST to retrieve data, you have to add X-HTTP-Method-Override:GET header.
If the earlier discussion around SMILES encoding all seems a bit too complicated (hence the xkcd comic strip) or just too much hassle, you can always use POST. But remember you won't be able to share SMILES links in your blog posts or Skype conversations :(
Is using POST to retrieve data 'breaking the web'?
There was a recent discussion about this topic after Dropbox team announced (just like us), that they see some limitations in GET compared to POST and will start to allow POST methods to retrieve data (original article). There were some critical opinions about allowing POST access to data, stating that this is a poor API design. This is turn has triggered a long discussion on hacker news page, from which one comment is particularity important: "I really like how the Google Translate API handles this issue. The actual HTTP method can be POST, but the intended HTTP method must always be GET (using the "X-HTTP-Method-Override" header)."
And this is exactly what we are doing, and we also believe this is the right way to use POST in order to allow retrieval of data from RESTful web interface.
 Today is election day in the UK, and so it's ones duty to vote, and then spend the rest of the day thinking about just how cool science is.I was thinking the other day on the train, that we don't always want drugs that are benign, of course we always want drugs that are safe and predictable in action; but mechanistically we sometimes want to do harm, kill a cell, wipe it out entirely - let's call these malign. Of course, this socially responsible expression of this desire to obliterate life with drugs is in the development of antibiotics (bacteria, viruses, fungi and parasites) and also anti-cancer agents. The other system we sometimes want to modulate to cause specific harm is via activation of the immune system - priming it to attack non-self (pathogen) cells, or that limbo-land of pseudo-self cancer cells.Anyway, this thinking of drugs as either (simplistically) malign or benign was triggered by a mail from a new collaborator interested in antiparasite agents identified via drug repositioning. How could you come up with a prioritised and annotated set of current drugs (and their associated cognate targets) that are biased to killing certain cells?
Today is election day in the UK, and so it's ones duty to vote, and then spend the rest of the day thinking about just how cool science is.I was thinking the other day on the train, that we don't always want drugs that are benign, of course we always want drugs that are safe and predictable in action; but mechanistically we sometimes want to do harm, kill a cell, wipe it out entirely - let's call these malign. Of course, this socially responsible expression of this desire to obliterate life with drugs is in the development of antibiotics (bacteria, viruses, fungi and parasites) and also anti-cancer agents. The other system we sometimes want to modulate to cause specific harm is via activation of the immune system - priming it to attack non-self (pathogen) cells, or that limbo-land of pseudo-self cancer cells.Anyway, this thinking of drugs as either (simplistically) malign or benign was triggered by a mail from a new collaborator interested in antiparasite agents identified via drug repositioning. How could you come up with a prioritised and annotated set of current drugs (and their associated cognate targets) that are biased to killing certain cells?



 As many of you know, SureChEMBL taps into the wealth of knowledge hidden in the patent documents. More specifically, SureChEMBL extracts and indexes chemistry from the full-text patent corpus (EPO, WIPO and USPTO; JPO titles and abstracts only) by means of automated text- and image-mining, on a daily basis. We have recently hosted a webinar about it which turned out to be very popular - for those who missed it, the video and slides are here.
As many of you know, SureChEMBL taps into the wealth of knowledge hidden in the patent documents. More specifically, SureChEMBL extracts and indexes chemistry from the full-text patent corpus (EPO, WIPO and USPTO; JPO titles and abstracts only) by means of automated text- and image-mining, on a daily basis. We have recently hosted a webinar about it which turned out to be very popular - for those who missed it, the video and slides are here.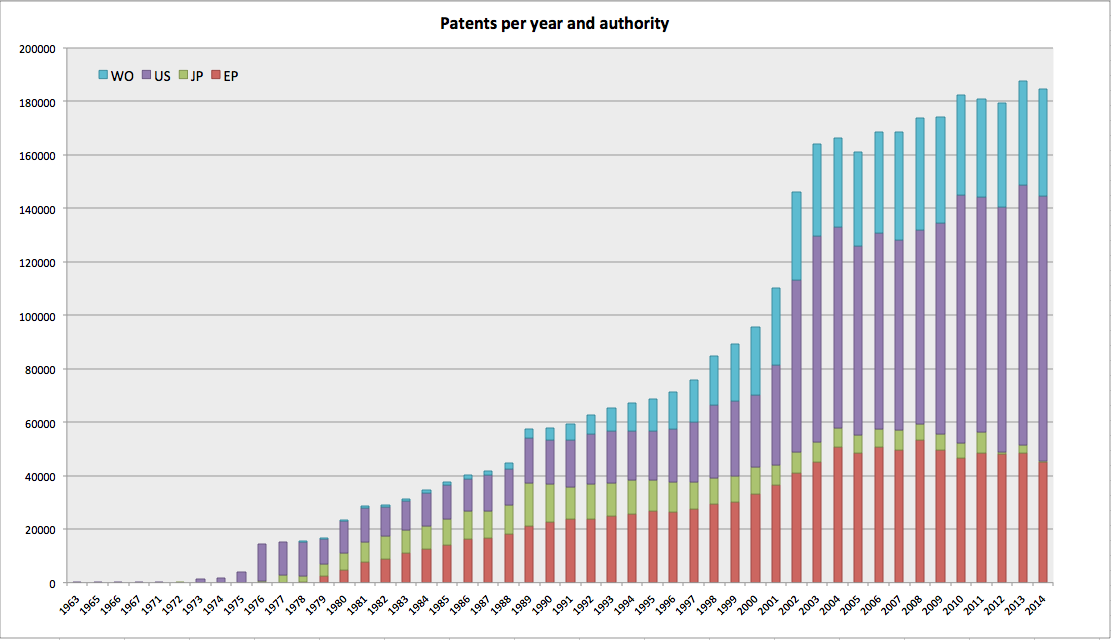
 We have mentioned Beaker (a.k.a the ChEMBL cheminformatics utility web service), several times on the blog (here, here and here), but have not devoted an entire post to Beaker. Well, here it is.
We have mentioned Beaker (a.k.a the ChEMBL cheminformatics utility web service), several times on the blog (here, here and here), but have not devoted an entire post to Beaker. Well, here it is.
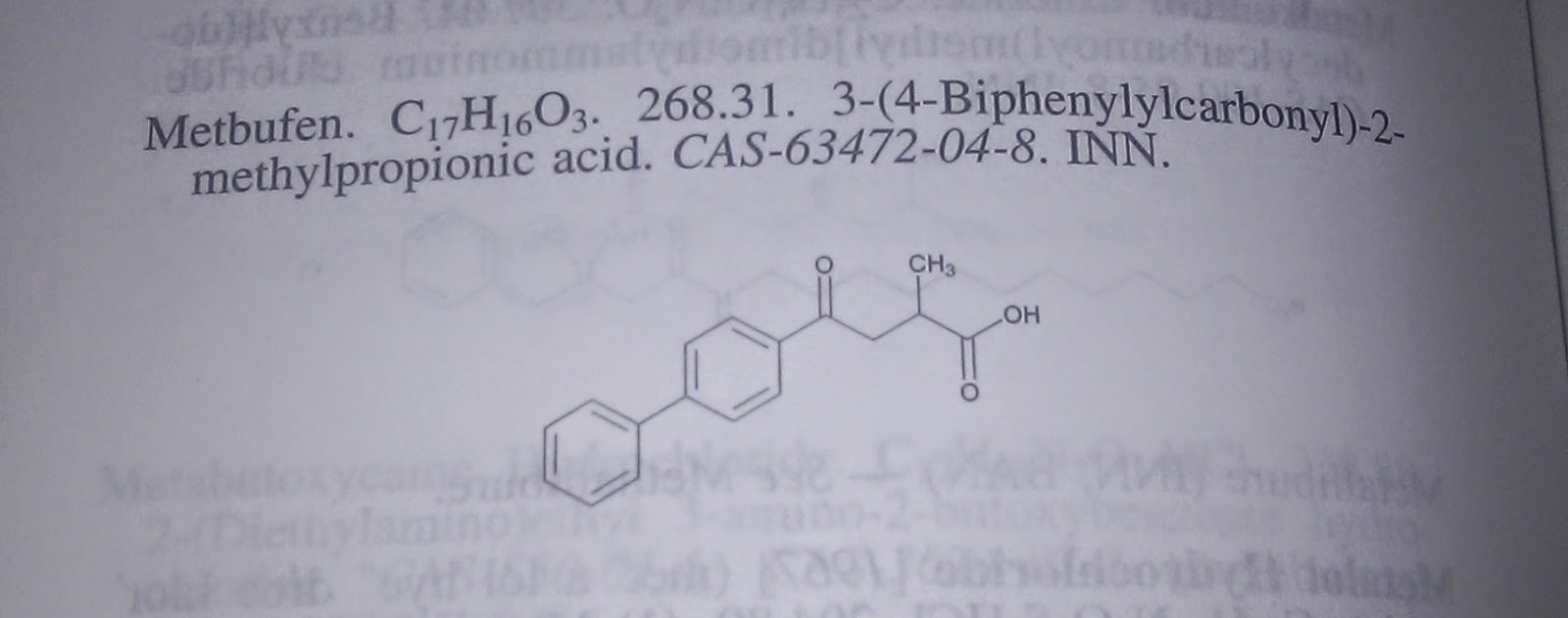 Original image available here.If we would like to find the compound in ChEMBL that is most similar to the one recognized from the image above, we could use this line of bash script:
Original image available here.If we would like to find the compound in ChEMBL that is most similar to the one recognized from the image above, we could use this line of bash script:


{kind=link}