-
Finding key compounds in med. chemistry patents: The open way

A couple of us attended the 3rd RDKit UGM, hosted by Merck in Darmstadt this year. It was an excellent opportunity to catch up with RDKit developments and applications and meet up with other loyal "RDKitters".
I presented a talk-torial there and went through an IPython Notebook, which some of you may find useful. It uses patent chemistry data extracted from SureChEMBL and after a series of filtering steps, it follows a few "traditional" chemoinformatics approaches with a set of claimed compounds. My ultimate aim was to identify "key compounds" in patents using compound information alone, inspired by papers such as this and this. The crucial difference is that these authors used commercial data and software, where in this implementation everything is free and open. At the same time, I wanted to show off what the combination of pandas, scikit-learn, mpld3, Beaker, RDKit, IPython Notebook and SureChEMBL can do nowadays (hint: a lot).
So, here is the Notebook and here are the associated slides which give a bit of background and context.Obviously, the logic and steps can be reimplemented with other toolkits or workflow tools, such as KNIME.
George
-
Using ChEMBL web services via proxy.

It is common practice for organizations and companies to make use of proxy servers to connect to services outside their network. This can cause problems for users of the ChEMBL web services who sit behind a proxy server. So to help those users who have asked, we provide the following quick guide, which demonstrates how to access ChEMBL web services via a proxy.
Most software libraries respect proxy settings from environmental variables. You can set the proxy variable once, normally HTTP_PROXY and then use that variable to set other related proxy environment variables:
Or if you have different proxies responsible for different protocols:
On Windows, this would be:
If you are accessing the ChEMBL web services programmatically and you prefer not to clutter your environment, you can consider adding the proxy settings to your scripts. Here are some python based recipes:
1. Official ChEMBL client library
If you are working in a python based environment, we recommend you to use our client library (chembl_webresource_client), for accessing ChEMBL web services. It already offers many advantages over accessing the ChEMBL web services directly and handling proxies is yet another. All you need to do is configure proxies once and you are done:
2. Python requests library
If you decide to use requests, you have to add 'proxies' parameter to every 'get' and 'post' function call:
3. Python urllib2 library
Finally, in the lowest level library, 'urllib2' you can set a ProxyHandler and register it to URL opener:
We would like to thank Dr. Christine Rudolph for the idea and providing code snippets. -
An overview and invitation to contribute to ChEMBL curation with PPDMs
PPDMs has been in the making for more than a year and is a follow-up on a conference paper we published in 2012. As in 2012, our objective is to map small molecule binding sites to protein domains, the structural units that form recurring building blocks in the evolution of proteins. An application note describing PPDMs is just out in Bioinformatics.
Mapping small molecule binding to protein domains
The mapping facilitates the functional interpretation of small molecule-protein interactions - if you understand which domain in a protein is targeted, you are in a better position to anticipate the downstream effect. Mapping small molecule binding to protein domains also provides a technical advantage to machine-learning approaches that incorporate protein sequence information as a descriptor to predict small molecule bioactivity. Reducing the sequence descriptor to the part that mediates small molecule binding increases the informative content of the descriptor. This is best exemplified by the domain-poisoning problem, illustrated below.

Result of a hypothetical query using as input the rat Tyrosine-protein phosphatase Syp (P35235) - and one of the hits, retrieved from a BLAST query against the ChEMBL target dictionary - the rat Tyrosine-protein kinase SYK (Q64725). The significant e-value for this query results from high scoring alignments of the SH2 domains. At the same time, the overlap between small molecules binding both proteins is expected to be low.
A simple heuristic
For individual experiments, it is often quite trivial to decide which domain was targeted. For example, medicinal chemists know whether their compound is a kinase inhibitor or one of a handful of SH2 inhibitors. This knowledge, while easily gleaned by the expert, is implicit and cannot be accessed programmatically. Hence we were motivated to implement a solution that could achieve this across as many measured bioactivities as possible.
Our initial implementation of mapping small molecules to protein domains consisted of a simple heuristic: Identify domains with known small molecule interaction and use these domains as a look-up when mapping measured bioactivities to protein domains. This process is illustrated in the figure below.
Despite its simplicity, this method works surprisingly well, owing to the fact that protein domains that are relevant to drug discovery are prioritised in Pfam-A model curation. Another factor that contributes here is the conservative route taken by many drug discovery projects that focus on targets that are in well characterised protein families. However, as illustrated by the cases labelled ii) and iii), some constellations are not covered by the simple heuristic.
A catalogue of validated domains was extracted from assays against single-domain proteins (step 1, 2) and projected onto measured bioactivities in ChEMBL (step 3). Three possible outcomes are: i) A successful mapping if exactly one of the Pfam-A domain models from the catalogue matches the sequence; ii) No mapping if none of the Pfam-A domain models from the catalogue match the sequence; iii) A conflicting mapping if multiple domain models from the catalogue match the sequence.
A public platform to review and improve mappings
Measured activities in ChEMBL falling into category iii) from the illustration above amount to only a fraction of the total but often reflect interesting biology. DHFR-TS for example is a multi-functional enzyme combining both a DHFR and Thymidylate_synt domain that occurs in the group of bikonts, which includes Trypanosoma and Plasmodium. In humans (and all metazoa), these domains occur as separate enzymes.
We built PPDMs as a platform to resolve such cases. PPDMs aggregates information that supports manual mapping assignments based on medicinal chemistry knowledge. New mappings can be committed to the PPDMs logs and then transferred to the ChEMBL database in future releases.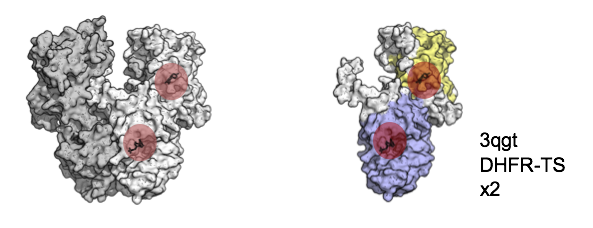
Small molecule inhibitors exist for both domains, DHFR (yellow, with Pyrimethamine) and Thymidylate synthase (blue, with Deoxyuridine monophosphate).
The Conflicts section on the website summarises conflicts (cases that correspond to category iii as discussed above) that were encountered when the mapping was applied to measured activities in the ChEMBL database and offers an interface to resolve them.
The Evidence section provides the full catalogue of domains for which we found evidence of small molecule binding. Evidence for the majority of domains in this list is provided in the form of measured bioactivities in ChEMBL, while in a few cases we provide a reference to the literature. These are cases where well-known domains occur exclusively in multi-domain architectures, such as 7tm_2 and 7tm_3. The catalogue can be downloaded in full from this section.
PPDMs also provides logs of individual assignments - these can be queried by date, user and comments left when the assignment was made. A log of all assigned mappings can be downloaded from this section. Another way to review assigned mappings is through the Resolved section, where assignments are grouped by domain architecture.
We invite everyone with an interest in the matter to sign up with PPDMs, whether it's simply for playing around, resolving remaining conflicts, or reviewing existing assignments. Please get in touch and we'll sort out a login for you!
felix -
Paper: PPDMs – A resource for mapping small molecule bioactivities from ChEMBL to Pfam-A protein domains
We've just published a Open Access paper in Bioinformatics on an approach to annotate the region of ligand binding within a target protein. This has a lot of applications in the use of ChEMBL, in particular providing greater accuracy in mapping functional effects, improving ligand-based target prediction approaches, and reducing false positives in sequence/target searching of ChEMBL. Where next for this work - well annotating to a site-specific level would be a good thing to implement (think about HIV-1 RT with the distinct nucleoside and non-nucleoside sites).
Here's the abstract...Summary: PPDMs is a resource that maps small molecule bioactivities to protein domains from the Pfam-A collection of protein families. Small molecule bioactivities mapped to protein domains add important precision to approaches that use protein sequence searches alignments to assist applications in computational drug discovery and systems and chemical biology. We have previously proposed a mapping heuristic for a subset of bioactivities stored in ChEMBL with the Pfam-A domain most likely to mediate small molecule binding. We have since refined this mapping using a manual procedure. Here, we present a resource that provides up-to-date mappings and the possibility to review assigned mappings as well as to participate in their assignment and curation. We also describe how mappings provided through the PPDMs resource are made accessible through the main schema of the ChEMBL database.
Availability: The PPDMs resource and curation interface is available at https://www.ebi.ac.uk/chembl/research/ppdms/pfam_maps
The source-code for PPDMs is available under the Apache license at https://github.com/chembl/pfam_maps.
Source code is available at https://github.com/chembl/pfam_map_loader to demonstrate the integration process with the main schema of ChEMBL. -
Django model describing ChEMBL database.

TL;DR: We have just open sourced our Django ORM Model, which describes the ChEMBL relational database schema. This means you no longer need to write another line of SQL code to interact with ChEMBL database. We think it is pretty cool and we are using it in the ChEMBL group to make our lives easier. Read on to find out more....
It is never a good idea to use SQL code directly in python. Let's see some basic examples explaining why:
Can you see what is wrong with the code above? SQL keyword `JOIN` was misspelled as 'JION'. But it's hard to find it quickly because most of code highlighters will apply Python syntax rules and ignore contents of strings. In our case the string is very important as it contains SQL statement.
The problem above can be easily solved using some simple Python SQL wrapper, such as edendb. This wrapper will provide set of functions to perform database operations for example 'select', 'insert', 'delete':
Now it's harder to make a typo in any of SQL keywords because they are exposed to python so IDE should warn you about mistake.
OK, time for something harder, can you find what's wrong here, assuming that this query is executed against chembl_19 schema:
Well, there are two errors: first of all `molecule_synonyms` table does not have a `synonims` column. The proper name is `synonyms`. Secondly, there is table name typo `molecule_synonyms`.
This kind of error is even harder to find because we are dealing with python and SQL code that is syntactically correct. The problem is semantic and in order to find it we need to have a good understanding of the underlying data model, in this case the chembl_19 schema. But the ChEMBL database schema is fairly complicated (341 columns spread over 52 tables), are we really supposed to know it all by heart? Let's leave this rhetorical question and proceed to third example: how to query for compounds containing the substructure represented by 'O=C(Oc1ccccc1C(=O)O)C' SMILES:
For Oracle this would be:
And for Postgres:
As you can see both queries are different, reasons for these differences are:
- Differences in Oracle and Postgres dialects
- Different chemical cartridges (Accelrys Direct and RDKit)
- Different names of auxiliary tables containing binary molecule objects
The example substructure search queries described above are similar to those used by the ChEMBL web services, which are available on EBI servers (Oracle backend) and in the myChEMBL VM (PostgreSQL backend). Still, the web services work without any change to their code. How?
All of the problems highlighted in this blogpost can be solved by the use of a technique known as Object Relational Mapping (ORM). ORM converts every table from database (for example 'molecule_dictionary') into Python class (MoleculeDictionary). Now it's easy to create a list of all available classes in Python module (by using 'dir' function) and check all available fields in class which corresponds to columns from SQL tables. This makes database programming easier and less error prone. The ORM also allows the code to work in a database agnostic manner and explains how we use the same codebase with Oracle and PostgreSQL backends.
If this blogpost has convinced you to give the ORM approach a try, please take a look at our ChEMBL example also included in myChEMBL:
-
myChEMBL 19 Released
 We are very pleased to announce that the latest myChEMBL release, based on the ChEMBL 19 database, is now available to download. In addition to the extra data, you will also find a number a great new features. So what's new then?
We are very pleased to announce that the latest myChEMBL release, based on the ChEMBL 19 database, is now available to download. In addition to the extra data, you will also find a number a great new features. So what's new then?More core chemoinformatics tools
We have included OSRA (Optical Structure Recognition), which is useful for extracting compound structures from images. OSRA can be accessed from the command line or by very convenient web interface, provided by Beaker (described below). We've also added OpenBabel - another great open source cheminformatics toolkit. This means you can now experiment with both RDKit and OpenBabel and use whichever you prefer.ChEMBL Beaker
myChEMBL now ships with a local instance the ChEMBL Beaker service. For those not familiar with Beaker, the service provides users with an array of chemoinformatics utilities via a RESTful API. Under the hood, Beaker is using RDKit and OSRA to carry out its methods. With the addition of Beaker in myChEMBL, users can now carry out the following tasks in secure local environment:- Convert chemical structure bewteen multiple formats
- Extract compound information from images and pdfs
- Generate compound images in raster (png) and vector (svg) forms
- Generate HTML5 ready representation of compound structure
- Generate compound fingerprints
- Generate compound descriptors
- Identify Maximum Common Substructure
- Compound standardisation
- Lots of more calculations
New IPython notebooks
We have written a number of new IPyhthon notebooks, which focus on a range of cheminformatics and bioinformatic topics. The topics covered by the new notebooks include:- Introduction on how to use ChEMBL Beaker
- Using the Django ORM to query the ChEMBL database
- Introduction to BLAST and creation of a simple Druggability Score
- Introduction to machine learning
- Analysis of SureChEMBL data, focused on identifying the MCS core identified in a patent
- Extraction and analysis of ChEMBL ADME data
We have also updated the underlying Ubuntu VM to 14.04 LTS, which also required us to make a number of changes the myChEMBL installation. To see how these changes and new additions have effected a bare metal installation of myChEMBL, head over the myChEMBL github repository.Installation
There are 2 different ways we recommend for installing myChEMBL:- Follow the instructions in the INSTALL file on the ftpsite. This will import the myChEMBL VM into VirtualBox
- Use Vagrant to install myChEMBL. See this earlier blogpost for more details, but the command to run is:
vagrant init chembl/myChEMBL && vagrant upIf you already have myChEMBL_18 installed via Vagrant, instead of running 'vagrant box update', we strongly recommend running:vagrant box remove chembl/myChEMBLvagrant init chembl/myChEMBL && vagrant upFuture plans
The myChEMBL resource is an evolving system and we are always looking to add new open source projects, tools and notebooks. We would be really interested to hear from users about what they would like to see in future myChEMBL releases, so please get in touch if you have any suggestions. (Just so you know, we already have a couple of ideas for myChEMBL 20).We hope you find this myChEMBL update useful and if you spot any issues or have any questions let us know.The myChEMBL Team -
New Drug Approvals 2014 - Pt. XII - Naloxegol (Movantik™)


ATC Code: A06AH03Wikipedia: NaloxegolChEMBL: CHEMBL2219418On September 16th FDA approved Movantik (naloxegol, AZ-13337019), as an oral treatment for patients with opioid-induced constipation and chronic non-cancer pain.
Naloxegol
Naloxegol is an opioid receptor antagonist. Due to its similarity to noroxymorphone, a main metabolite of oxycodone, naloxegol is classed as a controlled substance. However, the FDA analysed its abuse potential and concluded that there was no risk of dependency.

Naloxegol
Mode of ActionOpioids are a class of drugs which are used to manage pain, but have a common side effect of reducing the motility of the gastrointestinal tract, making bowel movements difficult. Opioids work by binding to the mu-receptors (CHEMBL233, UniProt:P35372) in the central nervous system, thereby reducing pain. However, they are also able to bind to the mu-receptors in the gastrointestinal tract, hence causing opioid-induced constipation.
Movantik is a peripherally-acting opioid receptor antagonist, which is able to prevent constipation by reducing this specific side effect of the opioids without affecting the efficacy of the pain management.
Clinical Trials
The clinical trials for this drug were carried out on a KODIAC clinical programme, comprising of four studies. Tests showed that 44% and 41% of patients receiving 25mg and 12.5mg, respectively, experienced increased bowel movements, compared to just 29% who took the placebo. [Paper]
Indication and WarningsThis drug is for non-cancer related pain. Side effects have been abdominal pain, diarrhea, headache and excessive gas in the stomach and/or intestinal area.
When used in conjunction with another peripherally-acting opioid antagonist, there is the chance of gastrointestinal perforation.
There is also the chance of withdrawal symptoms.
This is contraindicated for anyone who is also taking CYP3A4 (CHEMBL5792, UniProt:Q9HB55) inhibitors, such as clarithromycin (CHEMBL1741), as this will increase the exposure to naloxegol and could precipitate opioid withdrawal symptoms. [FDA]
Trade Names
Naloxegol was developed by AstraZeneca and is marketed under the trade name of Movantik. It is due for release during the first quarter of 2015. -
New Drug Approvals 2014 - Pt. XI - Idelalisib (Zydelig™)

ATC Code: L01XX47Wikipedia: IdelalisibChEMBL: CHEMBL2216870On July 23rd the FDA approved Zydelig (idelalisib, GS-1101), as an orally-delivered drug to treat patients with three types of blood cancers.
• Relapsed chronic lymphocytic leukemia (CLL)
• Relapsed follicular B-cell, non-Hodgkin lymphoma (FL)
• Relapsed small lymphocytic lymphoma (SLL)
Blood cancerThe three main categories of blood cancer are leukemia, lymphoma and myeloma. Lymphoma is also split into two types: Hodgkin lymphoma and non-Hodgkin lymphoma. Both leukemia and myeloma occur in the bone marrow, whilst lymphoma is a cancer that is isolated to the lymphatic system. Acute leukemia is where there is an abundance of underdeveloped white blood cells that can’t function properly and chronic leukemia is where there are just far too many white blood cells, which is just as bad as having too few. Myeloma is where the plasma cells form tumours in the bone marrow.
IdelalisibThis drug is a phosphoinositide 3-kinase inhibitor, which works by blocking P110σ (CHEMBL3130, Uniprot:O00329), the delta isoform of the phosphoinositide 3-kinase enzyme, encoded in humans by the PIK3CD gene. This isoform plays a role in B-cell development, proliferation and function and is expressed predominantly in leukocytes.

Idelalisib Mode of actionIdelalisib works on patients by inhibiting the PI3 kinase delta isoform (PI3Kδ), which plays an important role in malignant lymphocyte survival. It is the delta and gamma forms that are specific to the hematopoietic system. This treatment impairs the normal tracking of CLL lymph nodes. It can be used in conjunction with Rituxan (rituximab), an existing blood cancer treatment, for relapsed CLL and on its own for FL and SLL.
Clinical trialsClinical trials were carried out on 220 patients, with relapsed CLL, who were not healthy enough, due to co-existing medical conditions or damage from previous chemotherapy, to receive cytotoxic therapy. Patients were administered either idelalisib plus rituximab or a placebo and rituximab. Most of these patients were 65 years of age or older.After 24 weeks, 93% of the group who had taken the combination treatment were disease progression-free, compared to only 46% of the group who had received the placebo and rituximab combination.After 12 months, 90% of the dual drug combination group were alive, compared to 80% of the placebo-containing group. [NCI]
Indication and WarningsThis drug can be used in combination with rituximab or on its own, indicated for patients with relapsed conditions. There are several warnings for idelalisib, including hepatotoxicity, pneumonitis (fatal and serious), intestinal perforation and embyro-fetal toxicity. [FDA]
Trade NamesIdelalisib was developed by Gildead Sciences and is marketed under the name Zydelig.
