-
SureChEMBL Webinar
 As many of you know, SureChEMBL is one of our most recent resources, which taps into the wealth of knowledge hidden in the patent documents. More specifically, SureChEMBL extracts and indexes chemistry from the full-text patent corpus (EPO, WIPO and USPTO) by means of automated text- and image-mining, tirelessly, on a daily basis.If you would like learn more about SureChEMBL, its applications and exciting recent developments and future plans, we'll be giving a free webinar on Wednesday 11 March at 4pm GMT.
As many of you know, SureChEMBL is one of our most recent resources, which taps into the wealth of knowledge hidden in the patent documents. More specifically, SureChEMBL extracts and indexes chemistry from the full-text patent corpus (EPO, WIPO and USPTO) by means of automated text- and image-mining, tirelessly, on a daily basis.If you would like learn more about SureChEMBL, its applications and exciting recent developments and future plans, we'll be giving a free webinar on Wednesday 11 March at 4pm GMT.
Remember this is one of the times of year where daylight savings times may not be in sync, so check what time 4pm GMT is for your local timezone, for example, the time difference to Boston, MA at the moment is only 4 hours compared to the regular 5 hours.Please send us an email here to register your interest.George -
Using the New ChEMBL Web Services

As promised in our earlier post, here are some more details on making the most of the new ChEMBL web services. The best place to get started is to head over to the documentation page: https://www.ebi.ac.uk/chembl/api/data/docs. There you will find the list of resources (e.g. Molecule, Target and Assay) that are available and their methods. More importantly you can also execute each method with your own or default parameters, and view the URL, the response content and response status code. This is definitely the quickest way to start familiarizing yourself with the new ChEMBL web services.
Looking at the resources in more detail, you will find that each resource has three basic methods:
1. https://www.ebi.ac.uk/chembl/api/data/RESOURCE - will return all available objects of type RESOURCE from ChEMBL. An example could be https://www.ebi.ac.uk/chembl/api/data/molecule which returns all molecules (remember that data is paginated - more on this later).
2. https://www.ebi.ac.uk/chembl/api/data/RESOURCE/ID - will return a single object of type RESOURCE, identified by ID. For some resources, there can be more than one type of ID, for example the Molecule resource will accept:
- ChEMBL ID (https://www.ebi.ac.uk/chembl/api/data/molecule/CHEMBL1),
- SMILES (https://www.ebi.ac.uk/chembl/api/data/molecule/COc1ccc2%5BC@@H%5D3%5BC@H%5D%28COc2c1%29C%28C%29%28C%29OC4=C3C%28=O%29C%28=O%29C5=C4OC%28C%29%28C%29%5BC@@H%5D6COc7cc%28OC%29ccc7%5BC@H%5D56) and
- InChI key (https://www.ebi.ac.uk/chembl/api/data/molecule/GHBOEFUAGSHXPO-XZOTUCIWSA-N).
Note that all three examples above will return the same molecule. The same identifiers are valid for 'substructure' and 'similarity' resources.
Some resources can return more than one object for given ID, for example 'atc_class' will accept any ATC Level as it's ID. Only level 5 is unique across atc_class objects so https://www.ebi.ac.uk/chembl/api/data/atc_class/D05AX05 will return a single resource, but https://www.ebi.ac.uk/chembl/api/data/atc_class/D will return a list of dermatologicals.
3. https://www.ebi.ac.uk/chembl/api/data/RESOURCE/set/ID_1;ID_2;...;ID_N - will return N objects of type RESOURCE with IDs in set (ID_1, ID_2,...,ID_N), for example: https://www.ebi.ac.uk/chembl/api/data/molecule/set/CHEMBL900;CHEMBL1096643;CHEMBL1490 (note that ID separator is semicolon ';').
Format
If you do not specify a format, the default serialisation is XML. Other available formats include JSON and JSONP. There are three ways to provide format information:
- Extension, for example https://www.ebi.ac.uk/chembl/api/data/molecule/CHEMBL1.json
- Format parameter, for example https://www.ebi.ac.uk/chembl/api/data/molecule?format=json
- Accept header, for example:
For images the available formats are:
- png (https://www.ebi.ac.uk/chembl/api/data/image/CHEMBL1.png)
- svg (https://www.ebi.ac.uk/chembl/api/data/image/CHEMBL1.svg)
- json (https://www.ebi.ac.uk/chembl/api/data/image/CHEMBL1.json)
https://www.ebi.ac.uk/chembl/api/data/image/CHEMBL1.png?engine=indigo
Changing size works by adding 'dimensions' parameter:
https://www.ebi.ac.uk/chembl/api/data/image/CHEMBL1.png?engine=indigo&dimensions=100
The last thing worth noting about image format is that when you add X-Requested-With: XMLHttpRequest header to your request (i.e. you make an Ajax call), the resulting image will be base64-encoded. For example to make ajax call using jQuery library and render result as image you can use this code:
Alternatively, if you don't want to explicitly include the header in your jQuery code, you can use crossDomain: false parameter (yes, you are doing a cross-domain request, but we do support CORS so this will work as discussed here):
Pagination
When you make the following request https://www.ebi.ac.uk/chembl/api/data/molecule only the first 20 molecules will be returned. This corresponds to the first page of the molecule result set being requested. Pagination has been introduced to help reduce server load and also protect us from inadvertent DDoS attacks. It also allows clients to quickly obtain a portion of data without having to wait for the full data set. The most important page parameters are limit and offset, which are illustrated in the image below:

The red border presents a page of limit=10 and offset=10. Limit is a maximum number of objects on single page. Offset is a distance between the first element in result set and the first element in page. Please note, that objects are indexed staring from 0. The default limit is 20 and default offset is 0 and this is why accessing https://www.ebi.ac.uk/chembl/api/data/molecule provides first 20 elements. You can increase page size by providing bigger limit parameter, however the maximum allowed limit value is 1000.
All paginated results come in an 'envelope', which contains a resource object section and page metadata section. An example molecule page in json format looks like:
As can be seen, a 'page' of 20 molecule objects is stored in an array called 'molecules'. A 'page_meta' block provides information on page limit and offset. The 'page_meta' block also provides links (when available), to the previous and next pages and the total object count. In this example you can see that there are 1,463,270 compounds available in ChEMBL. The same information viewed in XML format:
Filtering
Filtering can be complex so let's start with the example. In the Molecule resource, there is a 'max_phase' numeric field. This is the maximum phase of development reached by a molecule. 4 is the highest phase and this means the molecule has been approved by the relevant regulatory body, such as the FDA. So let's select all approved drugs by adding a filter to the max_phase field:
https://www.ebi.ac.uk/chembl/api/data/molecule.json?max_phase=4
OK, so now we know that the filter can be passed as parameters and the simplest form is <field_name>=<value>, which means that we expect to get only items with field_name exactly matching the specified value. Right, let's add another filter. Inside a Molecule resource, we can find 'molecule_properties' object nested. One of the properties is a number of aromatic rings, so let's select compounds with at least two:
https://www.ebi.ac.uk/chembl/api/data/molecule.json?max_phase=4&molecule_properties__aromatic_rings__gte=2
Now we see, that many filters can be joined together using '&' sign. If the filter applies to the nested attribute we have to provide the name of the nested object first, followed by the name of the attribute, using double underscore '__' as a separator. Because we don't want to have an exact match, we have to explicitly specify the name of the relation, in our case 'greater then or equal' (gte). There are many other types of relations we can use in filters:
Filter TypesDescriptionExampleexact (iexact)Exact match with querycontains (icontains)Wild card search with querystartswith (istartswith)Starts with queryendswith (iendswith)Ends with queryregex (iregex)Regular expression querygt (gte)Greater than (or equal)lt (lte)Less than (or equal)rangeWithin a range of valuesinAppears within list of query valuesisnullField is null
Please note, that you can't use every relation for every type. For example regex matching is not allowed on numeric fields and ordering is forbidden on text. If you want to check, which filters can be applied to which resource, you can take a look at the resource schema, for example the 'molecule' resource, schema is available here:
https://www.ebi.ac.uk/chembl/api/data/molecule/schema.json
Ordering
Ordering is similar to filtering. As an example let's sort molecules by their molecular weight. There is a field called 'full_mwt' inside molecule properties, so ordering will look like this:
https://www.ebi.ac.uk/chembl/api/data/molecule.json?order_by=molecule_properties__full_mwt
And as we see, Helium is our first compound. In order to sort, we just have to add 'order_by' parameter with the value being the name of the field, prefixed with all intermediate nested objects. By default we sort ascending. To reverse this order and get molecules sorted from the heaviest to the lightest we have to add minus '-' sign before field name like this:
https://www.ebi.ac.uk/chembl/api/data/molecule.json?molecule_properties__isnull=false&order_by=-molecule_properties__full_mwt
This time the first element is a very heavy compound. Note that we had to add a filter to eliminate compounds without specified weight, otherwise, they will stick to the top of the results (This is because NULLS FIRST is the default for descending order in Oracle DB which we are using in production as described here). We can have multiple 'order_by' params in the URL like here:
https://www.ebi.ac.uk/chembl/api/data/molecule.json?order_by=molecule_properties__aromatic_rings&order_by=-molecule_properties__full_mwt
In which case molecules will be first sorted by the number of aromatic rings in ascending order, followed by molecular weight in descending order.
Filtering and ordering can be mixed together, but we leave it as an example for the reader.
Equivalent Web Service Requests
We will continue to support the 'old' web services until the end of the year. To help users with the upcoming migration process, the table below provides a mapping between the example web services requests found in the old documentation to the equivalent call in the new web services. It will be up to the end user to handle the different response format and the pagination of the returned data.
DescriptionOld Web Service URLNew Web Service URLCheck API statusGet compound by ChEMBLIDGet compound by Standard InChiKeyGet list of compounds matching Canonical SMILESGet list of compounds containing the substructure represented by a given Canonical SMILESGet list of compounds similar to the one represented by a given Canonical SMILES, at a given cutoff percentageGet image of a ChEMBL compound by ChEMBLIDGet individual compound bioactivitiesGet alternative compound forms (e.g. parent and salts) of a compoundGet mechanism of action details for compound (where compound is a drug)Get all targetsGet target by ChEMBLIDGet target by UniProt Accession IdentifierGet individual target bioactivitiesGet approved drugs for targetGet assay by ChEMBLIDGet individual assay bioactivities
ChEMBL Targets and UniProt Accessions
It is also worth noting that requests for ChEMBL targets by UniProt accessions is handled slightly differently in the new web services. It is now possible to return complexes, protein families and other target types, which contain the UniProt accession, not just the single protein targets. For example https://www.ebi.ac.uk/chembl/api/data/target?target_components__accession=Q13936&target_type=SINGLE%20PROTEIN, will return the ChEMBL target for Q13936. Removing the target_type filter, https://www.ebi.ac.uk/chembl/api/data/target?target_components__accession=Q13936, will return multiple targets (CHEMBL2095229, CHEMBL2363032 and CHEMBL1940 in ChEMBL release 20).
Expect some more web service related blog posts over the coming weeks and if you have any questions please get in touch.
The ChEMBL Team
-
New ChEMBL Web Services

Following on from our recent ChEMBL 20 release we are pleased to announce an updated version of the ChEMBL web services. First things first, some of the most important bits of information:
- You can explore new resources using online documentation available here: https://www.ebi.ac.uk/chembl/api/data/docs
- The code is Apache 2.0 licensed and available on GitHub: https://github.com/chembl/chembl_webservices_2
- The basic URL for accessing web services is https://www.ebi.ac.uk/chembl/api/data/ so in order to retrieve some information about molecules, you would construct following URL: https://www.ebi.ac.uk/chembl/api/data/molecule
- The 'old' web services are still available, so any applications and code relying on these services will continue to work. We do encourage you to review and migrate any code which uses these services to the updated versions described in this blog post.
And now some short Q&A session:
Q: But you already have web services available here: https://www.ebi.ac.uk/chemblws/ and they are sooo great, so why release something new?
A: With each new ChEMBL release more and more tables are being added to the underlying database schema, which have not been reflected in the web services for a long time. Additionally we had many suggestions from our users, which we have tried to address where possible. So in order expose more data and provide new functionality, it made sense to release the new and improved ChEMBL web services. The table below summaries the most important differences between old and updated ChEMBL web services:
FeatureOriginal ChEMBL Web ServicesUpdated ChEMBL Web ServicesBase URLhttps://www.ebi.ac.uk/chemblwshttps://www.ebi.ac.uk/chembl/api/dataNumber of resources517PaginationNoYesFilteringNoYesOrderingNoYesRaster ImagesYesYesVector ImagesNoYesJSONP supportYesYesCORS supportYesYesOnline DocumentationPython client libraryYesYesAvailable REST verbsGET, POSTGET, POSTSupport for SMILES in GETPartialFull
As you see, the most important difference are the number of new resources, for example we now include 'activity', 'cell_line', 'document' and many more. Existing resources (e.g. molecule, target, assay) have also been updated to include many new important attributes.
Another useful feature is filtering, you can now for example retrieve all molecules with a molecule weight less than 300 Da and preferred name ending with 'nib' using following URL:
https://www.ebi.ac.uk/chembl/api/data/molecule?molecule_properties__mw_freebase__lte=300&pref_name__iendswith=nib. You can apply filtering to every resource and filter by most of the fields belonging to given resource so for example targets that contain 'kinase' in pref_name would be: https://www.ebi.ac.uk/chembl/api/data/target?pref_name__contains=kinase. Don't worry if you do not follow the filtering query language, there will be a separate blog post on this topic.
Another new (and much requested) feature is the ability to easily retrieve multiple records from a resource. There are currently many ways to do this (filtering is one of them), but a more intuitive way is to get multiple records in bulk via the list of IDs. This is now possible, for example to retrieve ChEMBL molecules CHEMBL900, CHEMBL1096643 and CHEMBL1490, you can use: https://www.ebi.ac.uk/chembl/api/data/molecule/set/CHEMBL900;CHEMBL1096643;CHEMBL1490.
Q: Are the updated ChEMBL web services compatible with the old ones?
A: No. The URLs patterns and responses returned by the web services have changed, so they are not compatible. We recommend you review any code which currently consumes the old services and look to migrated to the updated version described here.
Q: But I have a lot of code that depends on the old web services! Will it break?
A: No. The old web services are still accessible and have been updated to serve data from ChEMBL 20 release. We will continue to support the old web services, but as part of our deprecation plan, we probably aim to stop supporting them by the end of the year. Please remember that the old code will always be available on GitHub and PyPI, so it is possible to create your own instance.
Q: How can I retrieve large data sets in the updated ChEMBL web services?
A: You can now retrieve all entities of a given resource, for example https://www.ebi.ac.uk/chembl/api/data/molecule returns all molecules in ChEMBL. This is achieved by returning the data in 'pages'. In order to implement paging, data is now wrapped in an envelope(<response> tag for XML output and outermost dictionary for JSON). Inside the envelope you can find collection of objects ('molecules' for molecule resource) and meta information looking like this:
Meta data provides information about current page: limit (how many objects can be on the page) and offset (serial number of the first object), as well as links to the previous and next page and total object count. Using the meta data it is now possible to loop through the large result set.
Q: Speaking about your python client library: will it include the old or the new version? Will it be backwards compatible?
A: Yes, version 0.8.x of the chembl_webresource_client package includes support for the updated web services. In order to access them, use following line of code:
If you list attributes of new_client object you will find that it contains all resources. The usage will be covered in separate blog post. We plan to drop support for the old services in version 0.9.x of the library, so the new_client will become default client.
Q: You claim that new web services supports GET and POST methods but online documentation lists only GET endpoints. I actually tried POST and I'm getting 405 Method now allowed error.
A: In the REST context GET, POST, PUT and DELETE are verbs meaning RETRIEVE, CREATE, UPDATE and DELETE accordingly. Because our web services are read-only we should only allow GET method. But GET has one very big limitation: all parameters (like identifiers, filtering, paging, ordering info) are appended to URL. All web servers (and some browsers) impose limits on URL length. In Apache, this limitation is 4000 characters by default. This means that if you want to get data about some (very) big molecule using SMILES as the identifier, it may happen that the resulting URL will be too long.
On the other hand, POST doesn't have this limitation as parameters are included in request body, not in URL. This is why we allow POST to retrieve data. But to be consistent with REST standard you have to let us now: 'OK, in this request I want to GET some data but I'm using POST just to pass more parameters'. In order to use it, just add header X-HTTP-Method-Override:GET to your POST request and you won't see 405 error anymore.
We hope you find the update useful and if you experience any problems or have any questions please get in touch.
The ChEMBL Team -
ChEMBL 20 Released

We are pleased to announce the release of ChEMBL_20. This version of the database was prepared on 14th January 2015 and contains:
- 1,715,135 compound records
- 1,463,270 compounds (of which 1,456,020 have mol files)
- 13,520,737 activities
- 1,148,942 assays
- 10,774 targets
- 59,610 source documents
Changes since the last release
In addition to the regular updates to the Scientific Literature, PubChem, FDA Orange Book and USP Dictionary of USAN and INN Investigational Drug Names this release of ChEMBL also includes the following new datasets:
AstraZeneca in-vitro DMPK and physicochemical properties
AstraZeneca have provided experimental data on a set of publicly disclosed compounds in the following ADMET related assays: pKa, lipophilicity (LogD7.4), aqueous solubility, plasma protein binding (human, rat, dog, mouse and guinea pig), intrinsic clearance (human liver microsomes, human and rat hepatocytes). For more details please refer to the AstraZeneca dataset document report page. Many thanks to Mark Wenlock and Nicholas Tomkinson for providing this data set.
MMV Malaria Box
Twelve new depositions from groups around the world screening the MMV Malaria Box have been loaded into ChEMBL 20. The groups include University of Yaoundé, University of Washington, University of Milan, Griffith University, Stanford University, National Cancer Institute, Weill Cornell Medical College, University Hospital Essen, Obihiro University of Agriculture and Veterinary Medicine, University of Toronto, Imperial College and Medicines for Malaria Venture.
HELM Notation
We have worked with members of the Pistoia Alliance to develop an implementation of the HELM standard for biotherapeutics and applied this to ChEMBL peptides (see associated press release). HELM notation has been generated for just under 19K peptides that were previously associated with Mol files in the database and that contain at least three amino acids. Our monomer library (containing the definitions/names of each of the amino-acids used) is available on the FTP site, and can be used by others to generate their own HELM notation. Abarelix is an example of a ChEMBL molecule that with HELM notation, which is now available on the compound report card page:

Structural Alerts
We have compiled a number of sets of publicly-available structural alerts where SMARTS were readily available and useable; these include Pfizer LINT filters, Glaxo Wellcome Hard Filters, Bristol-Myers Squibb HTS Deck Filters, NIH MLSMR Excluded Functionality Filters, University of Dundee NTD Screening Library Filters and Pan Assay Interference Compounds (PAINS) Filters. These sets of filters aim to identify compounds that could be problematic in a drug-discovery setting for various different reasons (e.g., substructural/functional group features that might be associated with toxicity or instability in in vivo info settings, compounds that might interfere with assays and for example, appear to be 'frequent hitters' in HTS).
It should be noted however that some alerts/alert sets are more permissive than others and may flag a large number of compounds. Results should therefore be interpreted with care, depending on the use-case, and not treated as a blanket filter (e.g., around 50% of approved drugs have 1 or more alerts from these pooled sets). The compound report card page now provides a summary count of the number of structural alerts hits picked up by a given molecule:
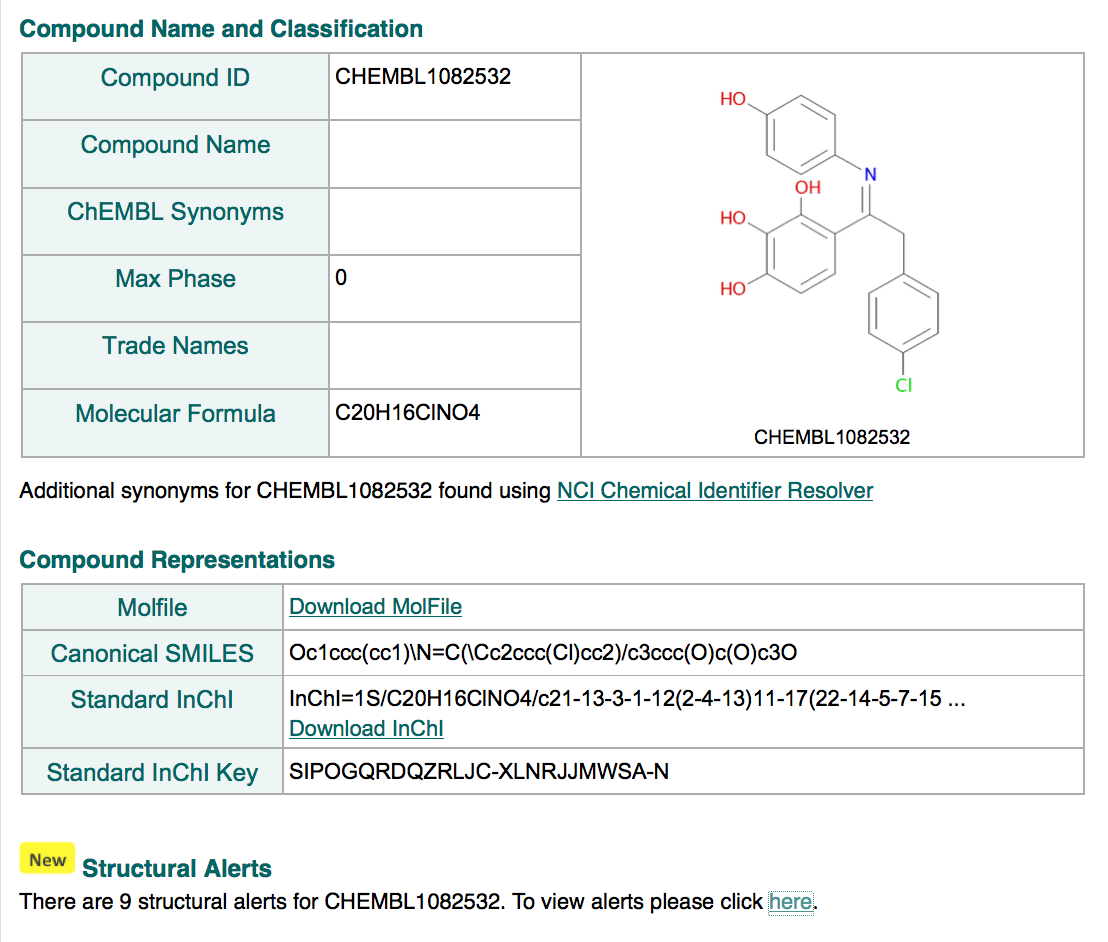
The link in the Structural Alert summary section takes the user to the Structural Alert Details page:

Pesticide MoA classification
For molecules in ChEMBL that are known pesticides, we have included the mode of action classification assigned by the Fungicide Resistance Action Committee (FRAC), Herbicide Resistance Action Committee (HRAC) and Insecticide Resistance Action Committee (IRAC). These classification schemes group pesticides both by their mode of action and chemical class. The classifications can be seen in the Compound Cross References section of the Compound Report Card pages. Thiabendazole is an example of a ChEMBL molecule which has been assigned a FRAC mode of action classification:
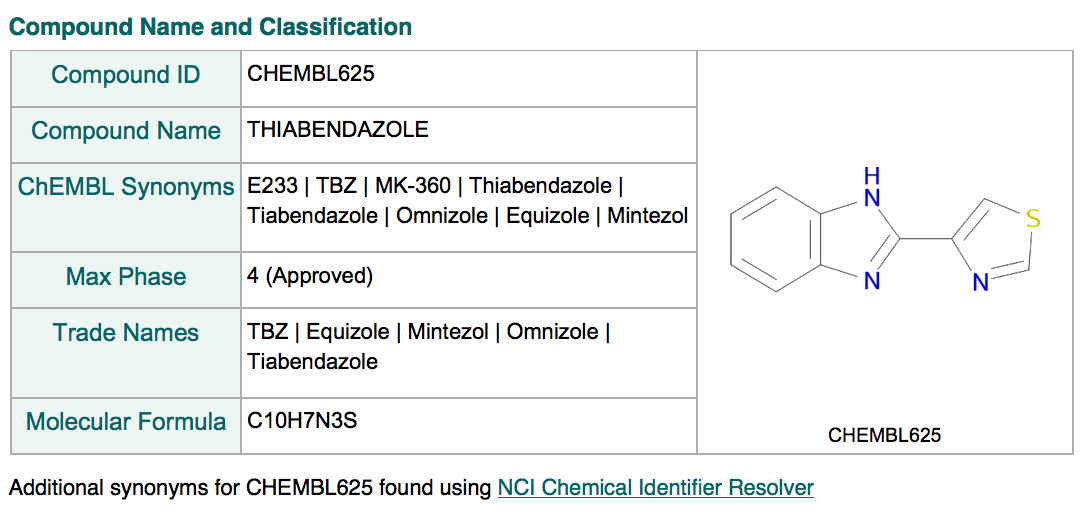
 This complements the ATC classification used for human drugs.
This complements the ATC classification used for human drugs.Cells, Cell Lines and LINCS integration
We now provide CHEMBL IDs for all cell lines stored in the ChEMBL database and we have also provided cross references to the LINCS project. To help users access the cell line data more easily we have setup a new cell search end point on the ChEMBL Interface, an example search output is displayed below:

A new Cell Report Card page has also been created:

EBI Complex Portal
For protein complex targets (e.g., CHEMBL2093869) we now have cross-references to the EMBL-EBI Complex Portal, a new resource providing manually curated information for stable protein complexes from key model organisms (see DOI: 10.1093/nar/gku975 for more details).MedChemComm content
As mentioned in the previous release, the visionary staff at the RSC have donated free access of their MedChemComm journal for abstraction into ChEMBL. This for us is a significant event, a commercial publisher giving access to their content, and assisting with it's extraction and integration into ChEMBL. So, here's a big thanks to them for this. Of course, this now sets us the challenge of selling this idea to other publishers!
RDF Update
The EBI-RDF Platform has also been updated with the ChEMBL 20 RDF. You can run the SPARQL queries online or download the ChEMBL 20 RDF files from the ftpsite.
We recommend you review the ChEMBL_20 release notes for a comprehensive overview of all updates and changes in ChEMBL 20 and as always, we greatly appreciate to reporting of any omissions or errors.
Keep an eye on the ChEMBL twitter and blog accounts for news on forthcoming myChEMBL and UniChem updates.
The ChEMBL Team -
The ChEMBL Roadshow: Part II
After the very successful US East Coast ChEMBL Roadshow, we (Anne and George) will be on the road once again next week to spread the word on ChEMBL, myChEMBL and SureChEMBL. This time we will visit the US West Coast and specifically these venues:- Tuesday 27th Jan: University of New Mexico, Albuquerque.
- Wednesday 28th: MolSoft/UCSD/Scripps, San Diego. See also here.
- Thursday 29th: IBM Research Centre, San Jose.
- Friday 30th: UCSF, San Francisco.
If you are nearby and would like to attend or meet us for a chat, please get in touch.We are grateful to SMSdrug.net for funding.George & Anne -
ChEMBL 20 schema
For those who just can't wait to update their code... Here is a picture of the new ChEMBL_20 schema.
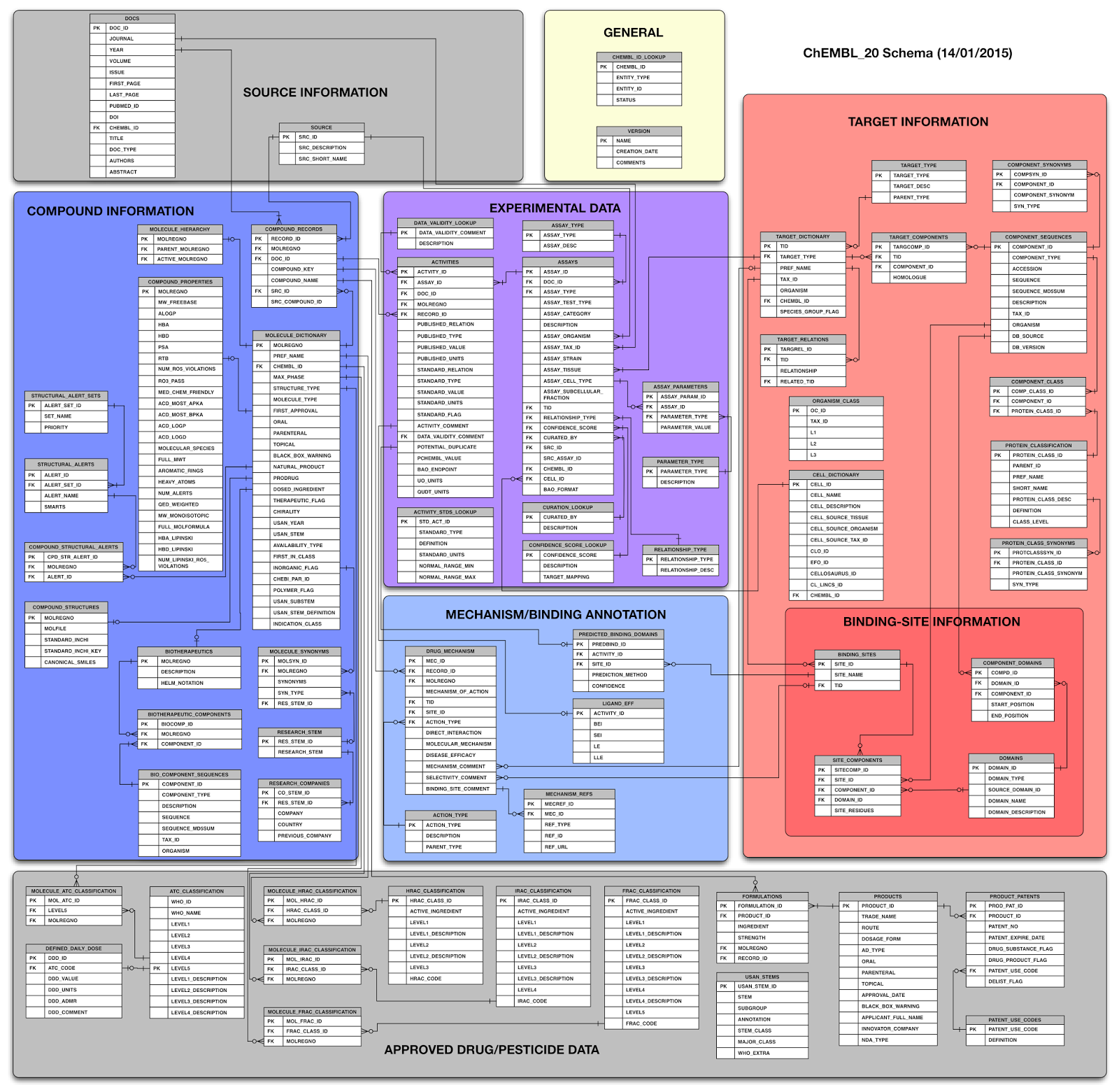
-
ChEMBL 20 coming soon...

Happy New Year for 2015 from the ChEMBL group!
Release 20 of the ChEMBL database will be happening around the end of the month, and for those who can't wait, here's a preview of the exciting new features you can expect to find there:
- HELM notation - we have developed an implementation of the Pistoia Alliance's HELM standard for biotherapeutics and will be supplying HELM notation for just under 20K peptides (previously represented by mol files). We will also make our monomer library available in case others wish to use it to generate their own HELM notation.
- Structural alerts - in place of the old 'Med Chem Friendly' flag used in ChEMBL, we now have an extensive set of structural alerts calculated for the ChEMBL compounds. The data set includes eight different sets of alerts (including sets published by Pfizer, Glaxo, BMS, University of Dundee, NIH MLSMR and PAINS filters) providing more than 1100 distinct SMARTS. Alerts found for a given compound can be viewed on the interface and the full data set will be included in the database downloads.
- Cell Report Cards - we will now provide ChEMBL IDs and Report Card pages for cells and cell-lines, as well as a keyword search facility. This will enable users to quickly identify all assays that have been performed in a particular cell-line and will also provide cross references to other resources such as the LINCS project.
- HRAC/FRAC/IRAC classification - for known herbicides, fungicides and insecticides, we provide their classification according to HRAC/FRAC/IRAC. This gives an indication of the mechanism of action of each compound. The classification can be found on the compound report card for these crop protection chemicals.
- New bioactivity data - in addition to the usual updates of bioactivity data from scientific literature and PubChem BioAssay we have 12 new sets of screening results from the MMV Malaria Box and an extensive set of in vitro DMPK and physiochemical property data for more than 5,700 publicly disclosed drugs and compounds deposited by AstraZeneca.
- There will be some schema changes to incorporate these new features, but these will be additions and are unlikely to break existing code.
The ChEMBL Team -
Accessing web services with cURL
ChEMBL web services are really friendly. We provide live online documentation, support for CORS and JSONP techniques to support web developers in creating their own web widgets. For Python developers, we provide dedicated client library as well as examples using the client and well known requests library in a form of ipython notebook. There are also examples for Java and Perl, you can find it here.
But this is nothing for real UNIX/Linux hackers. Real hackers use cURL. And there is a good reason to do so. cURL comes preinstalled on many Linux distributions as well as OSX. It follows Unix philosophy and can be joined with other tools using pipes. Finally, it can be used inside bash scripts which is very useful for automating tasks.
Unfortunately first experiences with cURL can be frustrating. For example, after studying cURL manual pages, one may think that following will return set of compounds in json format:
But the result is quite dissapointing...
The reason is that--data-urlencode (-d)tells our server (by setting Content-Type header) that this request parameters are encoded in "application/x-www-form-urlencoded" - the default Internet media type. In this format, each key-value pair is separated by an '&' character, and each key is separated from its value by an '=' character for example:
This is not the format we used. We provided our data in JSON format, so how do we tell the ChEMBL servers the format we are using? It turns out it is quite simple, we just need to specify a Content-Type header:
If we would like to omit the header, correct invocation would be:
OK, so request parameters can be encoded as key-value pairs (default) or JSON (header required). What about result format? Currently, ChEMBL web services support JSON and XML output formats. How do we choose the format we would like the results to be returned as? This can be done in three ways:
1. Default - if you don't do anything to indicate desired output format, XML will be assumed. So this:
will produce XML.
2. Format extension - you can append format extension (.xml or .json) to explicitly state your desired format:
will produce JSON.
3. `Accept` Header - this header specifies Content-Types that are acceptable for the response, so:
will produce JSON.
Enough boring stuff - Lets write a script!
Scripts can help us to automate repetitive tasks we have to perform. One example of such a task would be retrieving a batch of first 100 compounds (CHEMBL1 to CHEMBL100). This is very easy to code with bash using curl (Note the usage of the -s parameter, which prevents curl from printing out network and progress information):
Executing this script will return information about first 100 compounds in JSON format. But if you carefully inspect the returned output you will find that some compound identifiers don't exist in ChEMBL:
We need to add some error handling, for example checking if HTTP status code returned by server is equal to 200 (OK). Curl comes with--fail (-f)option, which tells it to return non-zero exit code if response is not valid. With this knowledge we can modify our script to add error handling:
OK, but the output still looks like a chaotic soup of strings and brackets, and is not very readable...
Usually we would use a classic trick to pretty print json - piping it through python:
But it won't work in our case:
Why? The reason is that python trick can pretty-print a single JSON document. And what we get as the output is a collection of JSON documents, each of which describes different compound and is written in separate line. Such a format is called Line Delimited JSON and is very useful and well known.
Anyway, we are data scientists after all so we know a plenty of other tools that can help. In this case the most useful is jq - "lightweight and flexible command-line JSON processor", kind of sed for JSON.
With jq it's very easy to pretty print our script output:
Great, so we finally can really see what we have returned from a server. Let's try to extract some data from our JSON collection, let it be chemblId and molecular weight:
Perfect, can we have both properties printed in one line and separated by tab? Yes, we can!
So now we can get the ranking of first 100 compounds sorted by their weight:
Exercises for readers:
1. Can you modify compounds.sh script to accept a range (first argument is start, second argument is length) of compounds?
2. Can you modify the script to read compound identifiers from a file?
3. Can you add a 'structure' parameter, which accepts a SMILES string. When this 'structure' parameter is present, the script will return similar compounds (you can decide on the similarity cut off or add an extra parameter)?


{kind=link}
{kind=link}
{kind=link}
{kind=link}