Congratulations to one of our collaborators, Paul Workman FMedSci of the Institute of Cancer Research on being awarded the RSC Chemistry World Entrepreneuur of the Year award for 2012. This award recognizes individuals that have made significant contributions to the commercialisation of science. Paul has of course got a fantastic record of significant scientific discovery in the area of molecularly targeted cancer research, but has consistently searched for ways of generating value and patient benefit from these by founding a number of companies to move these discoveries forward to potential therapies and products - spending time with Paul really makes clear the drive, belief and energy that's required to make a great scientific entrepreneur and innovator.
Examples of his commercialisation of science include the founding of Piramed (acquired in a successful exit by Roche) and Chroma Therapeutics.
So well done Paul! And for readers of the blog interested in hearing some of Paul's translational research, there's a great opportunity to do this at the EMBO Chemical Biology 2012 meeting.
On June 8th 2012, the US FDA approved pertuzumab (also known as RG-1273 and RhuMAb-2C4, tradename: Perjeta) for the treatment of HER2/ERBB2 positive, late stage metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease. Breast cancer is the most common female cancer. About 20% of breast cancers have amplified and over expressed Epidermal Growth Factor Receptor 2 (EGFR2, a.k.a. ERBB2 and HER2). These cancer subtypes are associated with worse prognosis and higher metastatic rates.
Pertuzumab is an anti-ERBB2/HER2 recombinant humanized monoclonal. It has been approved for use as part of a triple combination containing pertuzumab, another anti-ERBB2/HER2 antibody, trastuzumab, and the taxane docetaxel. The added value of combining both anti-ERBB2/HER2 antibodies is that pertuzumab binds to a different part of ERBB2 - the extracellular dimerization domain (Subdomain II) and this way it sterically blocks ligand-dependent heterodimerization with other HER family members. Meanwhile, trastuzumab binds to and inhibits the juxtamembrane portion of the extracellular domain.
Pertuzumab inhibits ligand-initiated intracellular signaling through the MAP kinase pathway, leading to cell growth arrest and the PI3 Kinase pathway, leading to apoptosis.
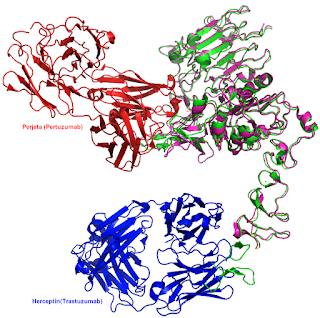
Superposition of the structures of pertuzumab (red) bound to ERBB2 (pink)- PDBe:1s78, with trastuzumab (blue) with ERBB2 (green) - PDBe:1n8z.
Pertuzumab has been issued a Black Box Warning because it can cause embryo-fetal death and birth defects, and thus cannot be used by women who are pregnant.
The target of pertuzumab is Human Epidermal Growth Factor Receptor 2 (ERBB2, HER2) (Uniprot:P04626; ; chembl:CHEMBL1824; canSAR:ERBB2-P04626).
There's a paper just published in Nature getting a lot of coverage on the internet at the moment from Novartis/UCSF, and for good reason - but as the cartoon above states, it will probably have less impact than news on Justin Bieber's new haircut, or the latest handbags from Christian Lacroix. It uses the SEA target prediction method, trained using ChEMBL bioactivity data in order to predict new targets (and then by association side effects) for existing drugs. These are then experimentally tested, and the results confirmed in a number of cases - this experimental validation is clearly complex and expensive, so it is great news that in silico methods can start to generate realistic and testable hypotheses for adverse drug reactions (there are also positive side effects too, and these are pretty interesting to look for using these methods as well).
The use of SEA as the target prediction method was inevitable given the authors involved, but following up on some presentations at this springs National ACS meeting in San Diego. There would also seem to be clear benefits in including other methods into linking a compound to a target - nearest neighbour using simple Tanimoto measures, and naive Bayes/ECFPP type approaches. The advantage of the SEA approach is that it seems to generalise better (sorry I can't remember who gave the talk on this), and so probably can make more comprehensive/complete predictions, and be less tied to the training data (in this case ChEMBL) - however as databases grow, these predictions will get a lot better. There will also be big improvements possible if other data adopts the same basic data model as ChEMBL (or something like the services in OpenPHACTS), so methods can pool across different data sources, including proprietary in-house data.
There are probably papers being written right now about a tournament/consensus multi-method approach to target prediction using an ensemble of the above methods. (If such a paper uses random forests, and I get asked to review it, it will be carefully stored in /dev/null) ;)
So some things I think are useful improvements to this sort of approach.
1) Inclusion of the functional assays from ChEMBL in predictions (i.e. don't tie oneself to a simple molecular target assay). The big problem here though is that although pooling of target bioassay data is straightforward - pooling/clustering of functional data is not.
2) Where do you set affinity thresholds, and how do the affinities related to the pharmacodyamics of the side-effects. My view is that there will be some interesting analyses of ChEMBL that maybe, just maybe, allow one to address this issue. Remember, we know quite a lot about the exposure of the human body, to a given drug at a given dose level...
3) Consideration of (active) metabolites. It's pretty straightforward now to predict structures of likely metabolites (not at a quantitative level though) and this may be useful in drugs that are extensively metabolised in vivo.
Anyway, finish off with some eye-candy, a picture from the paper (hopefully allowed under fair use!).
And here's a reference to the paper, in good old Bell AT&T labs refer format - Mendeley-Schmendeley as my mother used to say when I was a boy.
%T Large-scale prediction and testing of drug activity on side-effect targets
%A E. Lounkine
%A M.J. Keiser
%A S. Whitebread
%A D. Mikhailov
%A J. Hamon
%A J.L. Jenkins
%A P. Lavan
%A E. Weber
%A A.K. Doak
%A S. Côté
%A B.K. Shoichet
%A L. Urban
%J Nature
%D 2012
%O doi:10.1038/nature11159
Links to ChEMBL compounds from wikipedia have been there for some time, and now there is the target equivalent - for example here is the link to human thrombin.
We're looking for an intern for a six month project working with the great guys and gals of the 3DFrag project. The work will be based at the EBI, situated on a lovely science adjacent to the village of Hinxton near Cambridge (UK!), but the work will be performed in close collaboration with staff from the CRT Discovery labs in Cambridge.
The project will explore the relationship between chemical space and bioactivity space, using ChEMBL as a primary data source.
The aim would be to define chemical features which:
introduce large changes in bioactivity
relate to promiscuity against multiple targets
Based on the two properties, aim to select a set of fragments for screening which:
(a) Possess a high hit rate against particular target families
(b) Show high hit rates against a set of diverse targets
(c) Associated with being selective ligands.
Apart from being able to suggest fragment libraries for screening, an additional result of this work will be improved understanding of fragment space on the 'chemogenomics' level, i.e. to which extent targets (and target families) are related, based on experimentally determined fragment bioactivities.
The internship is associated with a tax-free stipend and can be arranged under a visa waiver program for suitable candidates. The group has access to a great computing facility and a wide range of software programs for cheminformatics, data analysis, and molecular modelling. The campus has a great canteen, and free transport from the centre of Cambridge and certain surrounding areas, accommodation is usually not a problem; nor is meeting a great crowd of fun people to spend spare time with!
We are looking for a cheminformatician/molecular modeller, ideally with experience on workflow tools such as Knime/PipelinePilot and experience of chemical structure handling and property calculation. Send us a cv/resume and we will review these and set up a phone interview with shortlisted candidates. The position is available immediately.
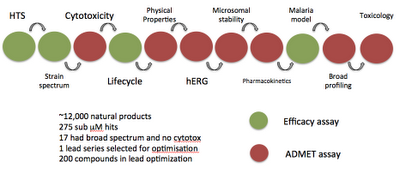
So, some more stuff on assays, in my quest to have something different to speak about over the summer; this post is about the tests a compound needs to pass through before it can become a drug. For a real test case, none of those green and red blobs I normally talk about, I took the excellent paper published in Science a few years ago - this is a great paper, discovering a clinical candidate for the treatment of malaria from a natural product screen. NITD609 is currently in phase 1 trials.
%T Spiroindolones, a Potent Compound Class for the Treatment of Malaria
%J Science
%D 2010
%V 329
%P 1175-1180
%O DOI: 10.1126/science.1193225
%A M. Rottmann
%A C. McNamara
%A B.K.S. Yeung
%A M.C.S. Lee
%A B. Zou
%A B. Russell
%A P. Seitz
%A D.M. Plouffe
%A N.V. Dharia
%A J. Tan
%A S.B. Cohen
%A K.R. Spencer
%A G.E. González-Páez
%A S.B. Lakshminarayana
%A A. Goh
%A R. Suwanarusk
%A T. Jegla
%A E.K. Schmitt
%A H.-P. Beck
%A R. Brun
%A F. Nosten
%A L. Renia
%A V. Dartois
%A T.H. Keller
%A D.A. Fidock
%A E. A. Winzeler
%A T.T. Diagana
The great thing in this paper is that it gives a reasonably complete package of data in the supplementary data, and from this it's possible to assemble the series of assays used to go from an HTS screen to a clinical development compound. I've put these together in the diagram below - as a linear graph. It's interesting to see that the majority of distinct assay types are connected with ADMET properties as opposed to efficacy. To be clear, this graph is one possible cascade of assays, formulated as a linear string, in reality, not all assays are done on all compounds, and some assays are done in parallel - but I'd still argue its a useful way to think about the progress of a compound to a drug (especially when this formulation is done at scale across many targets/diseases.
Another key point is that the ADMET assays are generic, i.e. they apply to essentially all drug discovery programs, and so can be happily abstracted out and treated separately (maybe ;) ).
Here's a diagram - I know, I know, it looks like it was done by a small child. Oh, and it is all about red and green blobs after all! Ways of improving it would be to have the numbers of input and output compounds at relevant stages, and maybe splitting out the lead discovery, from the lead optimisation assays (but the paper isn't that clear on this). Click image to make it bigger.
The order/paralellism aspect only affects the ADMET assays, the order of the efficacy assays will be as presented; my guess is, based on the systems I've looked at so far (not too many), that in general the efficacy assays will be linearly deployed, which has some good computational properties.
Here is a link to a Google Scholar page detailing papers that cite the NAR Database paper of Gaulton et al. We'll try and assemble these into an archive (for the Open Access ones only of course) somewhere into our plans on the new ChEMBL interface.








 Links to ChEMBL compounds from wikipedia have been there for some time, and now there is the target equivalent - for example here is the link to human thrombin.
Links to ChEMBL compounds from wikipedia have been there for some time, and now there is the target equivalent - for example here is the link to human thrombin.