-
Bioinformatics Training Course course KDMC11, July 12th - 15th, 2011, Portugal

There is an interesting course being run in Oeiras, Portugal in July.
About one hundred million different chemical compounds have already been synthesized. The number of theoretically possible organic molecules exceeds the number of atoms in the universe. This raises a number of questions, including:
- Where can one find information about chemical structures and their properties?
- How can one efficiently retrieve such information?
- Which molecules, if synthesized, could potentially assist the fight against certain types of cancer?
- Why is it that some pharmacological targets are considered more promising for the prevention or treatment of Alzheimer?s disease than others?
- Are there ways to better predict ADME(T) properties of synthesized molecules?
- Why does such a significant proportion of launched drugs originate from structures found in natural products?
- Why can multi-target pharmacological agents be superior to single-targeted ones?
- Can new medical applications be found for old drugs?
- Why is the alliance of chemo- and bioinformatics beneficial to the life sciences, biotech and pharma industries?
- What are the major challenges facing chemoinformatics now?
- Represent compounds and (bio)chemical reactions using chemical information in a computer.
- Search for information about chemical structures and their properties in public and commercially available databases.
- Perform similarity searches with an understanding of the advantages and disadvantages of the various methods.
- Prepare data sets for further (Q)SAR/(Q)SPR analysis, estimating the quality and completeness of the data.
- Create and validate (Q)SAR/(Q)SPR models for finding and optimization of lead compounds.
- Use the above techniques for virtual screening and design of chemical compounds with the required properties.
Researchers working in life sciences, professionals in the pharmaceutical and biotech industries: organic, medicinal, pharmaceutical chemists, biochemists, molecular biologists, pharmacologists, toxicologists, and others.
Information on all GTPB courses can be found at http://gtpb.igc.gulbenkian.pt -
GPCR Structures - Turkey beta-1 Receptor - further insight into antagonist-induced structural changes

Another clutch of turkey beta-1 adrenergic GPCR structures has recently been released (PDBe:2ycw, PDBe:2ycx, PDBe:2ycy and PDBe:2ycz), accompanying a publication in PNAS. These structures contain three different ligands (S-Carazolol, Cyanopindolol and Iodocyanopindalol), and sample new crystal forms, and reveal newly ordered regions not previously observed - these structures represent the fully inactivated R form of the receptor.
%T Two distinct conformations of helix 6 observed in antagonist-bound structures of a β1-adrenergic receptor %J Proc. Natl. Acad. Sci. USA %V 108 %P 8228–8232 %D 2011 %A R. Moukhametzianov %A T. Warne %A P.C. Edwards %A M.J. Serrano-Vega %A A.G.W. Leslie %A C.G. Tate %A G.F.X. Schertler
The annotated alignment and spreadsheet of GPCR structures has been updated. -
ChEMBL 10 Released
We are pleased to announce the release of ChEMBL_10. This latest version of the ChEMBL database contains:
- 1,118,566 compound records
- 1,000,468 distinct compounds
- 534,391 assays
- 4,668,202 bioactivities
- 8,372 targets
- 40,624 documents
- 6 data sources
- 333,864 compound records (PubChem Substance entries)
- 794 assays (PubChem BioAssay assays)
- 1,473,189 bioactivities (IC50 etc. measurements)
Changes to the interface include:
- 'Activity Source Filter' link has been added to the main search bar to allow users to include/exclude activity sources (e.g. PubChem BioAssay, Literature, ...) in the current working session
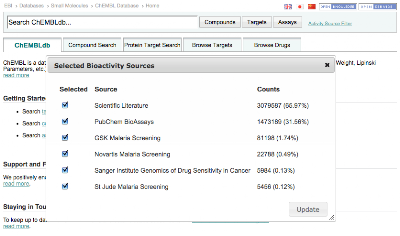
 You can download the data from the ChEMBL ftpsite: ftp://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/latest/
You can download the data from the ChEMBL ftpsite: ftp://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/latest/
All other ChEMBL resources (e.g. Web Services) are also now connected to ChEMBL_10. -
Safari plugin for ChEMBL data

For those of you who use the safari browser as part of your normal work, Matt Swain has written a plugin that you might find useful. You simply highlight words within a page, right-click, and then a box pops up with the option to search chembl for that chemical structure (using the selected text as the query). Matt also wrote similar plugins for ChemSpider, PubChem and OPSIN. These, and more, are available here. Thanks Matt!

-
Interested in Being at the Forefront of Text Mining for Drug Discovery Competitive Intelligence?

Well, so are we! If you are interested in applying for a postdoctoral fellowship in our group we would welcome you contacting us for further details. The position would apply text mining approaches across the broad internet to 'discover' interesting disclosures of either compound structures, progression through key clinical development or regulatory milestones, evidence of therapeutic efficacy for a compound/mechanism class etc. This data would be placed into the public domain, and integrated against other data sources, including ChEMBL, clinicaltrials.gov and other relevant data sources. You will need to have prior experience of entity recognition within free text, great scripting skills in a language such as perl or python, web analytics, and internet search engines (but hey, who doesn't know how to use google?). Additionally, experience of chemical structure extraction (image and/or text-based), ontology use/building and data integration and mining skills would be greatly beneficial.
The fellowship is intended for people wishing to pursue an independent research career, and significant autonomy and responsibility is available.
So, if you are interested, contact us. -
New Drug Approvals 2011 - Pt. XVII Fidaxomicin (Dificid TM)

ATC code (partial): A07AOn May 27th 2011, the FDA approved Fidaxomicin (Tradename: Dificid; Research Code: PAR-101, OPT-80, NDA 201699), a macrolide narrow spectrum antibacterial drug indicated for the treatment of Clostridium difficile-associated diarrhea (CDAD) in adults. Clostridium difficile (C. difficile) is an anaerobic, spore-forming Gram-positive bacteria, and overgrowth of this species can cause severe diarrhea and other more serious intestinal conditions, such as colitis.
Fidaxomicin is a fermentation product obtained from the Actinomycete Dactylosporangium aurantiacum. It exerts its therapeutic effect by inhibiting beta subunit of the bacterial enzyme DNA-directed RNA polymerase (RNAP) (UniProt:Q890N5), resulting in the death of C. difficile. Bacterial RNA polymerase is a large (~400 kDa) five subunit protein, and is the target of the already approved antibiotic rifampicin. Other treatments for CDAD already in the market include antibiotics such as Metronidazole (trade name Flagyl; ChEMBLID: CHEMBL137) and Vancomycin (ChEMBLID: CHEMBL262777). Patients generally respond to these antibiotic therapies, however there is a risk of recurrent infection associated with these treatments. Fidaxomicin has been shown to be more active in vitro than Vancomycin (minimum inhibitory concentration (MIC) of 0.12 µg/mL and 1.0 µg/mL, respectively) against C. difficile and also more selective, having limited activity in vitro and in vivo against components of the normal gut flora.
There are several known structures of bacterial RNA polymerases in complex with various antibiotics, typical is the structure of the Thermus aquaticus RNA polymerase in complex with sorangicin (PDBe:1ynn)
 The recommended dose of Fidaxomicin is one 200 mg tablet twice daily for 10 days (equivalent to a daily dose of 380 umol). At therapeutic doses, Fidaxomicin has a minimal systemic absorption, with plasma concentrations of Fidaxomicin and OP-1118, its main and microbiologically active metabolite, in the ng/mL range. The mean terminal half-life (T1/2) of Fidaxomicin and OP-1118 is 11.7 and 11.2 hours, respectively. Fidaxomicin is primarily transformed by hydrolysis at the isobutyryl ester to form OP-1118. Metabolism of Fidaxomicin and formation of OP-1118 are not dependent on cytochrome P450 (CYP) enzymes. Fidaxomicin is mainly excreted in feces, with 92% of the dose recovered as either Fidaxomicin and OP-1118.
The recommended dose of Fidaxomicin is one 200 mg tablet twice daily for 10 days (equivalent to a daily dose of 380 umol). At therapeutic doses, Fidaxomicin has a minimal systemic absorption, with plasma concentrations of Fidaxomicin and OP-1118, its main and microbiologically active metabolite, in the ng/mL range. The mean terminal half-life (T1/2) of Fidaxomicin and OP-1118 is 11.7 and 11.2 hours, respectively. Fidaxomicin is primarily transformed by hydrolysis at the isobutyryl ester to form OP-1118. Metabolism of Fidaxomicin and formation of OP-1118 are not dependent on cytochrome P450 (CYP) enzymes. Fidaxomicin is mainly excreted in feces, with 92% of the dose recovered as either Fidaxomicin and OP-1118. Fidaxomicin (IUPAC: [(2R,3S,4S,5S,6R)-6-[[(3E,5E,8S,9Z,11S,12R,13E,15E,18S)-12-[(2R,3S,4R,5S)-3,4-dihydroxy-6,6-dimethyl-5-(2-methylpropanoyloxy)oxan-2-yl]oxy-11-ethyl-8-hydroxy-18-[(1R)-1-hydroxyethyl]-9,13,15-trimethyl-2-oxo-1-oxacyclooctadeca-3,5,9,13,15-pentaen-3-yl]methoxy]-4-hydroxy-5-methoxy-2-methyloxan-3-yl]3,5-dichloro-2-ethyl-4,6-dihydroxybenzoate; SMILES: CC[C@H]1\C=C(/C)\[C@@H](O)C\C=C\C=C(/CO[C@H]2O[C@@H](C)[C@H](OC(=O)c3c(O)c(Cl)c(O)c(Cl)c3CC)[C@@H](O)[C@H]2OC)\C(=O)O[C@@H](C\C=C(/C)\C=C(/C)\[C@@H]1O[C@@H]4OC(C)(C)[C@@H](OC(=O)C(C)C)[C@H](O)[C@@H]4O)[C@H](C)O; ChEMBL: CHEMBL485861; PubChem: 46174142) has a molecular weight of 1058 Da, an ALogP of 7.7, seven hydrogen bond donors and 18 acceptors, and thus is not rule of five compliant. A notable feature is the 18-member polyene macrolide ring.
Fidaxomicin (IUPAC: [(2R,3S,4S,5S,6R)-6-[[(3E,5E,8S,9Z,11S,12R,13E,15E,18S)-12-[(2R,3S,4R,5S)-3,4-dihydroxy-6,6-dimethyl-5-(2-methylpropanoyloxy)oxan-2-yl]oxy-11-ethyl-8-hydroxy-18-[(1R)-1-hydroxyethyl]-9,13,15-trimethyl-2-oxo-1-oxacyclooctadeca-3,5,9,13,15-pentaen-3-yl]methoxy]-4-hydroxy-5-methoxy-2-methyloxan-3-yl]3,5-dichloro-2-ethyl-4,6-dihydroxybenzoate; SMILES: CC[C@H]1\C=C(/C)\[C@@H](O)C\C=C\C=C(/CO[C@H]2O[C@@H](C)[C@H](OC(=O)c3c(O)c(Cl)c(O)c(Cl)c3CC)[C@@H](O)[C@H]2OC)\C(=O)O[C@@H](C\C=C(/C)\C=C(/C)\[C@@H]1O[C@@H]4OC(C)(C)[C@@H](OC(=O)C(C)C)[C@H](O)[C@@H]4O)[C@H](C)O; ChEMBL: CHEMBL485861; PubChem: 46174142) has a molecular weight of 1058 Da, an ALogP of 7.7, seven hydrogen bond donors and 18 acceptors, and thus is not rule of five compliant. A notable feature is the 18-member polyene macrolide ring.
The full prescribing information can be found here.
The license holder for Fidaxomicin is Optimer Pharmaceuticals, Inc. and the product website is www.dificid.com. -
How do I get a reasonably complete, reasonably accurate list of PPIs?

A recurring thing we are interested in is classifying the natural ligand/partner of a protein. Having a classification of the 'natural' ligand partner for every human protein would be really useful, and would allow us to ask and more importantly answer quite complex questions in ChEMBL like - 'show me all small molecules that modulate a protein-protein interaction (PPI) at better than 100 nM potency?'.
I've had a bit of a dig around the web, but there isn't anything that does exactly what I need (or I'm not smart enough to find it), but I'm sure it must exist, so in the comments section, please post resources and ideas..... -
New Drug Approvals 2011 - Pt. XVI Telaprevir (IncivekTM)

ATC code (partial): J
On May 23rd, the FDA approved Telaprevir (Tradename: Incivek; Research Code: VX-950, NDA 201917), a Hepatitis C virus NS3 protease (HCV NS3) inhibitor, for the treatment of chronic hepatitis C virus genotype 1 infection, in combination with peginterferon alfa and ribavirin.
HCV is a prolonged infection that affects the liver and is caused by a small positive single-stranded RNA virus, which is transmitted by blood-to-blood contact. Chronic hepatitis C is normally asymptomatic, but may lead to liver fibrosis, and if untreated, potentially fatal liver failure. There is currently no vaccine for this type of hepatitis.
Telaprevir is an inhibitor of the hepatitis C virus (HCV) non-structural protein 3 (NS3) protease (ChEMBLID:CHEMBL4893; Uniprot ID:A3EZI9), a viral protein required for the proteolytic cleavage of the HCV encoded polyprotein (UniProt:P27958) into mature forms of the NS4A, NS4B, NS5A and NS5B proteins (NS3 is Uniprot: P27958[1027-1657]). These proteins are involved in the formation of the virus replication complex, and therefore are vital to its proliferation. In a biochemical assay, Telaprevir inhibited the proteolytic activity of the recombinant HCV NS3 protease domain with an IC50 value of 10 nM.
HCV NS3 is a serine protease (Pfam:PF02907). There are many protein structures known for this protein in complex with inhibitors, a typical entry is PDBe:3rc4, as expected from early genome annotation, the NS3 proteinase has a fold distantly related to the chymotrypsin-like family of serine proteinases, and contains the classic Asp-His-Ser catalytic triad.
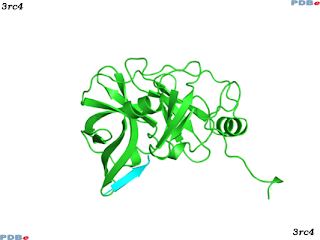
The -vir USAN/INN stem covers antiviral agents, and the substem -previr indicates it is a serine protease inhibitor. Telaprevir is the second approved agent to target HCV NS3, following the approval earlier this month of Merck's Boceprevir (q.v.). Other compounds in this class in late stage clinical development/registration include Tibotec's TMC-435, and Bristol Myers Squibb's Asunaprevir (BMS-650032). Others at earlier stages of development include ABT-450, BI-201335, IDX-320, MK-5172, Vaniprevir (MK-7009), Narlaprevir (SCH-900518), Danoprevir (RG-7227, ITMN-191), BIT-225, VX-500, ACH-1625, GS-9256.

Telaprevir (IUPAC: (1S,3aR,6aS)-2-[(2S)-2-({(2S)-2-cyclohexyl-2-[(pyrazin-2-ylcarbonyl)amino]acetyl}amino)-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrole-1-carboxamide; SMILES: CCCC(C(=O)C(=O)NC1CC1)NC(=O)C2C3CCCC3CN2C(=O)C(C(C)(C)C)NC(=O)C(C4CCCCC4)NC(=O)C5=NC=CN=C5; PubChem:3010818; ChEMBL ID: CHEMBL231813) has a molecular weight of 679.8 Da, contains 4 hydrogen bond donors, 8 hydrogen bond acceptors, and has an ALogP of 2.69. The inhibitor is clearly peptide like, containing four amino acid residues, mimicking the natural substrate of the protease, and including a 'warhead' - the alpha-keto amide, which covalently binds to the catalytic serine residue of the target enzyme.
Telaprevir is available as oral film-coated tablets of 375 mg. It has an apparent volume of distribution (Vd/F) of approximately 252 L, and, in patients who received a dose of 750 mg three times a day (the recommended daily dose is therefore a large 2.25 g (equivalent to 3,310 umol)), the exposure is characterised by an AUC of 22,300 ng.hr/mL, with a Cmax of 3510 ng/mL. Telaprevir should be administered with a standard fatty meal, since its bioavailability is enhanced by 237%. In vitro, protein plasma binding ranges from 59% to 76%.
The predominant metabolites of Telaprevir in plasma are the R-diastereoisomer (VRT-127394), which is approximately 30-fold less potent than the parent drug, pyrazinoic acid, and a metabolite that underwent reduction of the α-ketoamide bond of Telaprevir (which is, as expected not active against the target). Telaprevir is also metabolised by CYP3A4, being simultaneously a substrate and an inhibitor, and therefore, other therapeutic agents metabolised by CYP3A4 may prolong their therapeutic effect or adverse reactions. See prescribing information for the extensive list of drug-drug interactions and contraindications.
Following administration of a single oral dose of 750 mg, Telaprevir is eliminated with a mean plasma half-life (t1/2) of approximately 4.0 to 4.7 hours, and it has a mean total body clearance (CL/F) of approximately 32.4 L/hr.
Telaprevir was developed almost in parallel with Boceprevir, the first-in-class inhibitor of the HCV NS3 protease. Both drugs require a high daily dose for an effective response, and are generally similar with respect to their pharmacokinetic and pharmacokinetic parameters.
The license holder for Telaprevir is Vertex Pharmaceuticals Incorporated, and the full prescribing information can be found here. For more information, please visit the product website here.











{kind=link}